
Abstract
Idiosyncratic drug reactions can be extremely severe and are not accounted for by the regular pharmacology of a drug. Thus, the mechanism of idiosyncratic drug–induced liver injury (iDILI), a phenomenon that occurs with many drugs including β-lactams, anti-tuberculosis drugs and non-steroidal anti-inflammatories, has been difficult to determine and remains a pressing issue for patients and drug companies. Evidence has shown that iDILI is multifactorial and multifaceted, which suggests that multiple cellular mechanisms may be involved. However, a common initiating event has been proposed to be the formation of reactive drug metabolites and covalently bound adducts. Although the fate of these metabolites are unclear, recent evidence has shown a possible link between iDILI and the adaptive immune system. This review highlights the role of reactive metabolites, the recent genetic innovations which have provided molecular targets for iDILI, and the current literature which suggests an immunological basis for iDILI.
What is iDILI?
Idiosyncratic drug–induced liver injury (iDILI) describes an adverse liver reaction that is not attributed to the normal pharmacological action of that drug and is a function of the biology of the individual patient. The mechanisms of iDILI are still poorly understood but are believed to be multistep and multicellular in nature. The incidence of these reactions is extremely low and so iDILI is difficult, if not impossible, to predict during preclinical trials. 1,2 Symptoms can manifest as hepatic, cholestatic or mixed injury with severe elevations in alanine aminotransferase (ALT) levels. Often, the consequence is acute liver failure and, ultimately, patient morbidity and mortality. 3,4
Many different classes of drugs may cause iDILI, but the most common are antibiotics and non-steroidal anti-inflammatories, namely co-amoxiclav, flucloxacillin, diclofenac and isoniazid. 5 –9 It therefore seems unlikely, given the mixed clinical picture and the different classes of drugs involved, that a single mechanism is responsible for iDILI. 10,11 Moreover, similar symptoms may be the result of different mechanisms even within the same class of drugs. 12 Among the suggested mechanisms of DILI are tumour necrosis factor α–induced apoptosis, mitochondrial dysfunction, cell stress, genetic polymorphisms, viral infection and activation of the innate and adaptive immune systems. 13 –19
This review highlights the action of reactive drug metabolites as a molecular initiating event in iDILI, the increasing importance of using genetic polymorphisms to understand phenotypic mechanisms and the role of the adaptive immune system in iDILI. Three drugs will then be used to illustrate how these factors interact to cause iDILI, namely isoniazid, co-amoxiclav and flucloxacillin.
The role of CRMs
Whilst liver metabolism is able to convert drugs into inert compounds for excretion, in many instances, chemically reactive drug metabolites (CRMs) are formed. 20 Metabolic profiling of candidate drugs during development can be used to exclude drugs that form harmful toxicophores through metabolism. But, unfortunately, not all reactive metabolites may be identified during discovery or the activity level of the metabolite may be below the lower limit of detection. 21 Advances in the sensitivity of mass spectrometry and in CRM screening may also change over the course of drug development. 22 Whilst all the mechanisms that lead from CRM formation to the onset of iDILI are still unclear, two major pathways that are involved include direct cell toxicity and cell damage leading to immune activation.
Most CRMs are electrophilic species which have the ability to conjugate with proteins or other macromolecules in their vicinity. Their biological targets are difficult to determine and are largely dependent on the stability of the CRM and its half-life. 23 An increase in CRMs will deplete protective mechanisms in the liver, such as glutathione, which are responsible for removing reactive oxidative species. 24 Antioxidative genes and their proteins may also be activated via the nuclear factor (erythroid-derived 2)-like 2 protective pathway. 15 If these protective mechanisms fail, then CRM toxicity could form covalently bound conjugates leading to protein dysfunction, altered regulation of drug transporters, mitochondrial toxicity and lysosomal perturbations. 25 –27 When enough of these cell components are affected, then cell death is likely. For example, the inhibition of bile acid pumps can result in the intracellular accumulation of a cytotoxic bile acid. 28 As many transporters are energy dependent, defects in transporters may show linkage with mitochondrial toxicity. 26
In each of these proposed mechanisms, the initiating adverse event is the formation of a CRM. However, each mechanism alone may not be sufficient to trigger liver damage to the extent that is often seen in iDILI. Additionally, the quantity of covalent binding in some studies does not show a direct correlation with the incidence of iDILI. 29 The multiple determinant hypothesis suggests that iDILI is dependent on the build-up of many different factors that increase the probability of an adverse event. 30 Whilst CRM formation and covalent binding are some of these factors, they may also be part of a series of events that lead to immune activation. These pathways are demonstrated in Figure 1. The hypothesis also states that one of these factors must be the presence of a genetic association. The recent expansion in genomic studies may show this to be true.

A schematic to demonstrate the pathways that lead to cell death in drug-induced liver injury due to direct toxicity and immune-mediated toxicity. Importantly, within an environment of cell stress, danger signalling to the immune system can occur.
Genetic advances in iDILI
Common genetic associations among iDILI patients have helped identify some of the components involved in liver toxicity (Table 1). Among the first associations to be identified were allele polymorphisms in metabolizing enzymes including the cytochrome P450 oxidizing system, N-acetyl transferases and glucoronosyl transferases. Other associations include drug transporters and superoxide dismutase. 16,51 Diclofenac, a non-steroidal anti-inflammatory drug, can form highly toxic CRMs under the metabolism of CYP2C8 and also UDP-glucuronosyltransferase-2B7 (UGT2B7). 35,52 Additionally, polymorphisms in the transporter allele, adenosine triphosphate-binding cassette, sub-family C (ABCC2) have also been associated with susceptibility to iDILI in diclofenac-treated patients. 35
Examples of established genetic associations with iDILI drugs.
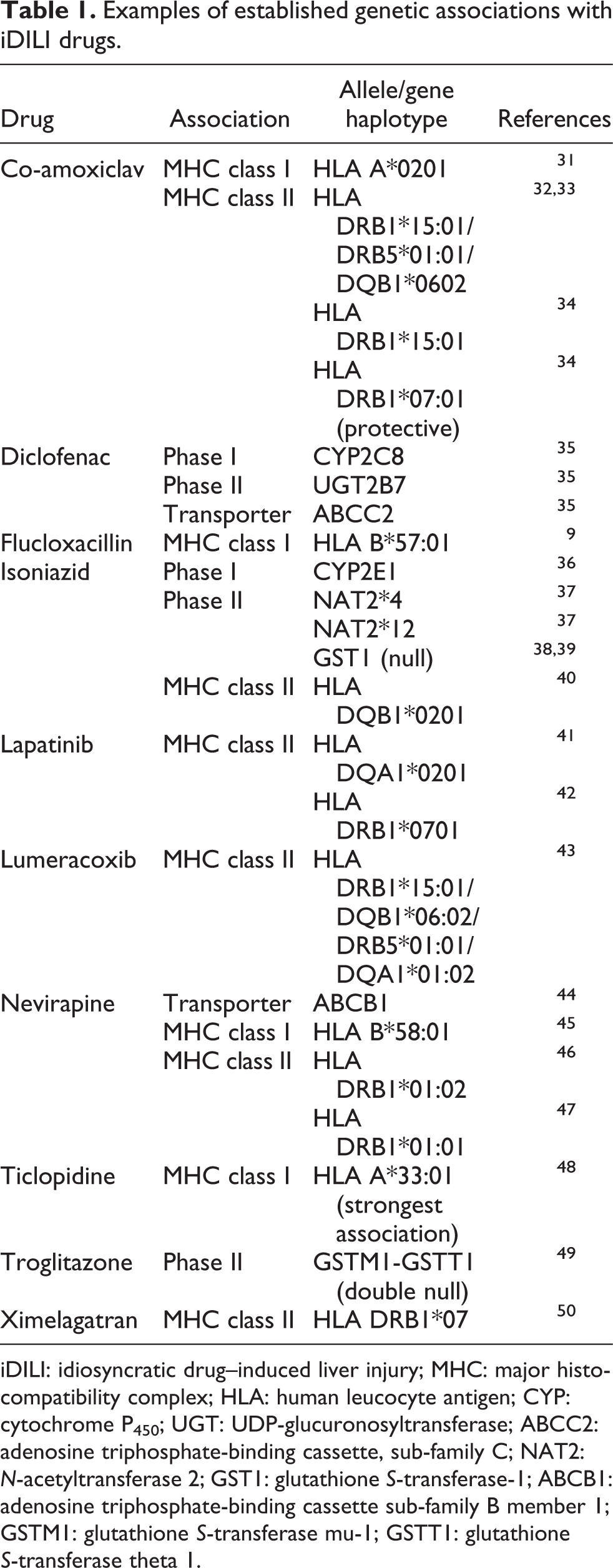
iDILI: idiosyncratic drug–induced liver injury; MHC: major histocompatibility complex; HLA: human leucocyte antigen; CYP: cytochrome P450; UGT: UDP-glucuronosyltransferase; ABCC2: adenosine triphosphate-binding cassette, sub-family C; NAT2: N-acetyltransferase 2; GST1: glutathione S-transferase-1; ABCB1: adenosine triphosphate-binding cassette sub-family B member 1; GSTM1: glutathione S-transferase mu-1; GSTT1: glutathione S-transferase theta 1.
A significant number of human leucocyte antigen (HLA) associations have been found with drugs that cause iDILI (Table 1) and they are increasingly being shown to have a high negative predictive value in a clinical setting. 53 This implies that that the adaptive immune system may be important in the disease aetiology. 10 A strong association between the HLA B*57:01 allele and the HIV therapeutic, abacavir, has led to an understanding of the critical role of T-cells in abacavir hypersensitivity. 45,54 The mechanism for abacavir hypersensitivity has now been deduced and the discovery has resulted in a functional change in clinical practice. 55 Although abacavir is not a culprit for iDILI, a similar association has been found with flucloxacillin and HLA B*57:01. 9 The link with iDILI and HLA makes a strong case for an immunological mechanism for iDILI.
More recently, multiple gene polymorphisms have shown to appear together and may have a mechanistic link. For example, when the UGT2B7 and ABCC2 polymorphisms were considered together, they showed a stronger association with diclofenac hepatotoxicity. Although, no mechanistic links have yet been found. 35 With the rise of bioinformatics, we may be able to identify genetic haplotypes and the relevant biological components involved.
Immune mechanisms of liver injury
The formation of CRMs, covalent binding and genetic polymorphisms can, in theory, all contribute to iDILI. However, there may be other factors contributing to an immune-mediated drug reaction. The evidence that supports an immunological aetiology for iDILI includes the delayed onset where symptoms appear only several weeks after the administration of the drug, 56,57 an increase in susceptibility to iDILI where immune activation has already occurred due to viral infection, 18 the presence of T-cells in liver biopsies from patients with iDILI 58,59 and the isolation of drug-specific T-cells in patients with iDILI. 10 The liver is an immune-privileged organ so that the mechanisms that maintain the tolerogenic micro-environment must be overcome for immune activation to occur. 60
The role of T-cells in delayed-type drug hypersensitivity reactions involving the skin 61 are much better understood, but the mechanism of immune activation remains the same. Two factors are critical for immune activation to occur: maturation of antigen-presenting cells and presentation of novel antigens to T-cells. The action of CRMs may cause liver cells to release danger signals, which may result in the activation of antigen-presenting cells. 62 Cells that are under stress or undergoing necrosis will release damage-associated molecular patterns and pro-inflammatory cytokines. 63 Both are involved in the maturation of antigen-presenting cells and their recruitment to the area of damage. 64 Under the influence of these ‘danger signals’, co-stimulatory molecules are upregulated, which increase their ability to activate T-cells. The hapten model suggests that the CRM will bind covalently to endogenous proteins to form protein adducts. Whilst a drug molecule could be too small to be deemed immunologically relevant, a protein adduct could undergo antigen processing by antigen-presenting cells and be presented by major histocompatibility complex (MHC) molecules and recognized by a T-cell as a foreign antigen. 65 This pathway is also highlighted in Figure 1. Investigative studies on three drugs can link the formation of drug protein adducts and their genetic associations with immune-mediated iDILI.
Isoniazid
Development of iDILI due to anti-tuberculosis therapy has shown a strong genetic link with two polymorphisms in the drug-metabolizing enzyme N-acetyltransferase 2 (NAT2). 37 Presence of NAT2*4 and NAT2*12 results in slow acetylation of the drug which is associated with an increased risk of hepatotoxicity 66,67 due to greater accumulation of the acetylhydrazine CRM in these patients. 68 Another genetic association is with the HLA DQB1*0201 allele, which suggests an immunological aetiology possibly due to the presence of CRMs. 40,69 In one instance, lymphocyte transformation tests were performed in 95 patients with iDILI to show a drug-specific memory T-cell response in 26% of these patients. Furthermore, this was increased to 56% of patients upon the addition of a prostaglandin inhibitor to cultures. 70 These comprehensive studies were based on initial studies performed by Warrington et al. with isoniazid, where the lymphocyte transformation test was initially proposed as a way of identifying immune activation in hepatotoxicity. 71,72 More recently, anti-isoniazid antibodies were found in patient sera alongside anti-CYP450–isoniazid adduct antibodies. These were not found in isoniazid-treated controls, which suggests that if a humoral response is detected, adduct formation must be involved. 73 Finally, we have recently isolated T-cells from patients with iDILI to generate isoniazid-specific T-cell clones (unpublished data).
Co-amoxiclav
Co-amoxiclav is a combined therapy containing two different β-lactam antibiotics, amoxicillin and clavulanic acid. β-Lactams are more commonly associated with drug hypersensitivity reactions involving the skin due to the formation of protein adducts. 74,75 Whilst amoxicillin alone does not commonly cause iDILI, it does cause iDILI when given in combination with clavulanic acid. 76 A role for the adaptive immune system is also supported by studies linking co-amoxiclav hypersensitivity with a number of HLA class II alleles, particularly the HLA DRB1*15:01 and HLA DQB1*06:02 haplotype. 11,31,34 One study has linked specific haplotypes with specific clinical symptoms of iDILI: the DRB1*15:01/DQB1*06:02 haplotype with cholestatic iDILI and the HLA A*30:02/HLA B*18:01 haplotype with hepatocellular iDILI. 33 Furthermore, drug-specific T-cell clones have been isolated from co-amoxiclav–treated patients with iDILI. Several pieces of evidence indicate that the clones were activated via a hapten mechanism implicating chemically reactive haptens in the aetiology of iDILI (Kim et al., 2015). 77 It is important to note that T-cell clones isolated from patients were not cross-reactive and were specific for either amoxicillin or clavulanic acid.
Flucloxacillin
Flucloxacillin-induced liver injury is strongly associated with HLA B*57:01. 9 Drug-specific T-cell clones have been isolated from patients and have been fully characterized. 10 Mainly CD8+ T-cells were isolated with the cytotoxic capacity to cause cell damage. These cells also express liver homing receptors CCR4 and CCR9. This suggests that T-cells with the ability to cause cell death could, in theory, be recruited to the liver. Importantly, flucloxacillin-specific T-cells were restricted by HLA B*57:01 and required antigen processing to generate an antigen to trigger a T-cell response. Another study comparing patient and volunteer clones showed that patients clones responded to a haptenic drug antigen, requiring antigen processing, whereas volunteer clones could be stimulated by drug directly without antigen processing. 78 Like other β-lactam antibiotics, flucloxacillin binds to lysine residues on proteins, and these protein conjugates have been discovered and quantified in vitro. 79 Most recently, CD8+ T-cells were found in a liver biopsy, and flucloxacillin-specific T-cells, generated in vitro from normal volunteers carrying HLA B*57:01, were shown to kill liver cell lines transfected with HLA B*57:01. 59 Exposure of experimental animals to flucloxacillin may also result in the activation of naive CD8+ T-cells. Drug treatment ex vivo resulted in the secretion of interferon γ and granzyme B and the induction of hepatocyte apoptosis. 80 A key issue in iDILI is identifying the cellular and molecular site of antigen formation responsible for generating drug-specific T-cells capable of causing liver damage. Animal models have suggested that flucloxacillin may indeed generate haptens in the liver, 81 but an essential area of ongoing research is to define the molecular and cellular interactions in the liver that are the first steps in liver injury.
A future direction for iDILI
As yet there are no generally accepted assays that allow for the prediction of iDILI during drug development. This is partly because we have only a rudimentary understanding of the mechanisms involved in iDILI for the majority of drugs involved but also because immunological mechanisms are difficult to detect in high throughput analysis. A clear strategy is needed to link allele polymorphisms with aetiological mechanisms.
Mass spectrometry has improved our ability to profile CRMs in liver microsomal incubations and hepatocyte models. However, determining the fate of a metabolite in vitro is difficult because it has the potential to interact with numerous biological targets. For example, the drug ticlopidine is known to form a number of different metabolites in vitro, 82 but this research alone has done little to further our understanding of whether these CRMs are responsible for iDILI. However, when put in light of its strong association with HLA A*33:03, a number of investigative avenues become clear. 48 For example, naive T-cells from volunteers expressing this allele can be primed to ticlopidine in order to investigate the nature of the drug–antigen in vitro. 74,83
Unfortunately, in vitro models cannot reproduce all the complex pathways involved in iDILI. Hence, there is an urgent requirement to fully elucidate the mechanisms involved in animal models with iDILI. Recent work with the antimalarial amodiaquine has made an important step forward in animal models for iDILI using PD-1 (−/−) mice to model liver injury. However, much more extensive studies with other drugs are needed. 84,85
A variety of different genetic polymorphisms associated with iDILI suggests the involvement of a wide variety of toxicological mechanisms. However, the number of the HLA associations discovered and the recent data on T-cells implies that immunological mechanisms play a critical role in the aetiology of iDILI.
Footnotes
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
AT is a PhD student funded by GSK.