
Abstract
The aims of this study were to assess the utility of the PXB mouse model of a chimeric human/mouse liver in studying human-specific effects of an important human hepatotoxic drug, the PPARγ agonist, troglitazone. When given orally by gavage for 7 days, at dose levels of 300 and 600 ppm, troglitazone induced specific changes in the human hepatocytes of the chimeric liver without an effect on the murine hepatic portions. The human hepatocytes, in the vehicle-treated PXB mouse, showed an accumulation of electron-dense lipid droplets that appeared as clear vacuoles under the light microscope in H&E-stained sections. Following dosing with troglitazone, there was a loss of the large lipid droplets in the human hepatocytes, a decrease in the amount of lipid as observed in frozen sections of liver stained by Oil-red-O, and a decrease in the expression of two bile acid transporters, BSEP and MRP2. None of these changes were observed in the murine remnants of the chimeric liver. No changes were observed in the expression of three CYPs, CYP 3A2, CYP 1A1, and CYP 2B1, in either the human or murine hepatocytes, even though the baseline expression of the enzymes differed significantly between the two hepatocyte species with the mouse hepatocytes consistently showing increased expression of the protein of all three enzymes. This study has shown that the human hepatocytes, in the PXB chimeric mouse liver, retain an essentially normal phenotype in the mouse liver and, the albeit limited CYP enzymes studied show a more human, rather than a murine, expression pattern. In line with this conclusion, the study has shown a differential response of the human versus the mouse hepatocytes, and the effects observed are highly suggestive of a differential handling of the compound by the two hepatocyte species although the exact reasons are not as yet clear. The PXB chimeric mouse system therefore holds the clear potential to explore human hepatic–specific features, such as metabolism, prior to dosing human subjects, and as such should have considerable utility in drug discovery and development.
Introduction
Troglitazone was the first of a class of thiazolidinedione drugs registered for the treatment of type 2 diabetes in 1997 (Yokoi 2010). While the drug proved very effective in reducing glycemia in patients with type 2 diabetes, it was withdrawn in the United States in 2000 because of the appearance of serious, and sometimes fatal, idiosyncratic liver injury in its clinical use (Kohlroser et al. 2000; Watkins and Whitcomb 1998). The histological findings of troglitazone-induced liver injury in man included hepatocyte necrosis, bile duct proliferation, and cholestatic hepatitis (Gitlin et al. 1998; Neuschwander-Tetri et al. 1998). While troglitazone was, and still is, being extensively studied in animals, the exact initiating events for hepatotoxicity in man remain essentially unknown, while the nonclinical toxicology profile was relatively benign with little suggestion of the hepatotoxicity that subsequently developed in the clinic, albeit in a small proportion of those taking the drug (Roth and Ganey 2010; Watkins 2005). One proposed mode of action is the production of protein-reactive metabolites by human hepatic drug metabolizing systems that are missing or reduced in the common preclinical species used, that bind to, and alter/damage, essential macromolecules in the cells (Madsen et al. 2008). Troglitazone has also been shown to be a potent inhibitor of the drug transporter systems in both rat and human hepatocytes in vitro (Jemnitz et al. 2010; Snow and Mosely 2007), and this could lead to cholestasis and subsequent liver disease in a susceptible population of patients.
The comparative insensitivity of preclinical testing for this type of human liver damage has prompted various investigators to explore manipulation of the preclinical test system to better predict the outcome in man, in an attempt to avoid the dangerous, and potentially avoidable, appearance of hepatic injury in the clinic. Hence, approaches to increase the sensitivity of preclinical test systems have included pretreatment of rodents with inducers of hepatic inflammation, such as bacterial lipopolysaccharide (Roth and Ganey 2010), by the rational use of isolated hepatocytes in screening cascades in vitro (Stieger, Meier, and Meier 2007), by the use of isolated membrane vesicles overexpressing relevant hepatic drug transporters such as BSEP and MRP2 (Hayashi et al. 2005), by cell lines specifically expressing human hepatic drug metabolizing enzymes (THLE-CYP cell lines; Greer et al. 2010; Mace et al. 1997), and by the in silico prediction of the possible generation of reactive chemical species (Park et al. 2005; Vignati et al. 2005). While each of these approaches has their advantages, none of them has been found to truly recapitulate the in vivo environment present in an intact human being undergoing an idiosyncratic, drug-induced, adverse event.
In an attempt to address the problem of laboratory animal to human extrapolation, several in vivo chimeric human/mouse liver models have been recently developed based upon the urokinase-type plasminogen activator–transgenic SCID mice (uPA/SCID mice; Barth et al. 2008; Yoshizato, Tateno, and Utoh 2011). These mice, known as PXB mice, in the absence of a human hepatic repopulation of the mouse liver, undergo a progressive liver failure (Tateno et al. 2004), which rapidly reverses upon infusion of isolated preparations of human hepatocytes into the spleen, and the subsequent replacement of the murine components of the rapidly failing mouse liver with fully functional human hepatocytes. The PXB mouse model was specifically developed as a means of establishing a population of human hepatocytes in a mouse liver to potentially allow an experimental way of directly addressing the human metabolism of chemicals without actually having to expose humans to potentially toxic chemical species (Sato et al. 2008).
The aims of the present study were therefore to investigate the potential use of the PXB mouse in helping to characterize the underlying hepatic effects of troglitazone, given by oral gavage, to the mouse. In order to achieve this, the biological characteristics of the untreated, and troglitazone-treated, chimeric liver were studied in terms of the their expression of certain forms of cytochrome P450 and also that of two canalicular membrane transporters involved in bile acid regulation, the bile salt export pump (BSEP) and the multidrug resistance–associated protein 2 (MRP2; Funk et al. 2001b; Horikawa et al. 2003).
Materials and Methods
Chemicals
Sodium hypochlorite was supplied by Wako Pure Chemical Industries Ltd. (Osaka, Japan). Isoflurane (FORANE®) anesthesia was obtained from Abbott Japan Co. Ltd. (Tokyo, Japan). Troglitazone (batch 46) and an authentic metabolite standard, troglitazone sulfate (batch EN00350-012-01), was synthesized by AstraZeneca Sweden (Molndal, Sweden) and had a chemical purity >98%.
Animals
SCID mice were sourced from PhoenixBio Co. Ltd (Higashi–Hiroshima, Japan). Chimeric PXB mice with humanized livers were generated at PhoenixBio Co. Ltd. by the method described previously (Tateno et al. 2004). Cryopreserved human hepatocytes (Lot Number BD85; male African American; 5 years old) were purchased from BD Biosciences (San Jose, CA). These were transplanted into the spleen of 2- to 4-week-old male and female uPA+/+/SCID mice under diethyl ether anesthesia. After transplantation, male mice with a blood human albumin concentration of greater than 7.0 mg/mL were selected for dosing. On the day prior to administration, 2 µL of blood was collected from the tail vein of each PXB mouse, diluted in 200 µL of LX buffer EIKEN (Eiken Chemical Co. Ltd., Tokyo, Japan), and centrifuged (400g; 10 min, 4°C). The human albumin concentration was measured from these samples based on latex agglutination immunophelometry as described by Ritchie (1975). All mice used in the study were aged 10 to 14 weeks and weighed more than 16 g prior to dosing. Mice were housed individually in cages (175 × 245 × 125 mm3) and provided with food (solid pellet; CRF-1 containing Vitamin C; Oriental Yeast Co. Ltd., Tokyo, Japan) and sterilized public tap water containing sodium hypochlorite ad libitum. The animal room was maintained at 20°C to 26°C with 42 to 72% relative humidity and a 12-hour light–dark cycle. All animals were randomized into dosing groups based on their mean values for body weight and human albumin to minimize the variance between the groups.
All animals were housed, maintained, and handled according to international standards of animal health and welfare in an AAALAC (The Association for Assessment and Accreditation of Laboratory Animal Care International)-accredited facility, and all experimental procedures were performed with the approval of the ethics board of the Hiroshima Prefectural Institute of Industrial Science and Technology, Hiroshima, Japan.
Dosing
Groups of SCID and PXB mice (3/group) were dosed by oral gavage with troglitazone in 0.5% methylcellulose (Sigma Chemical Co., St Louis, MO) at dose levels of 0, 300, and 600 mg/kg/day for 7 days. Analysis of dosing preparations confirmed that the intended concentrations of the test article were achieved.
Clinical Biochemistry
Blood was sampled and analyzed for various markers of hepatic function as described and reported in Schulz-Utermohl et al (2011). This data will not be reported in this publication.
Necropsy and Tissue Preparations
Animals were sacrificed by cardiac puncture and exsanguination, after anesthesia with isoflurane. Whole livers were harvested and weighed at necropsy. A liver slice of 5 mm in thickness was taken from the left lobe and fixed in formalin. The remaining liver tissue was snap frozen in liquid nitrogen. The formalin-fixed liver samples were kept at room temperature for 24 hours, dehydrated in a graded series of ethanol (70%, 80%, 90%, and 100%), infiltrated with xylene, embedded in paraffin, and stored at room temperature until sectioning and analysis by H&E staining and immunohistochemistry (IHC) for specific proteins (details given below). Frozen tissue samples were used for Oil-red-O staining and for subsequent analysis by electron microscopy by methods described in the appropriate sections.
IHC
IHC was used to identify both human- and mouse-specific proteins within the liver sections and to study the relative expression of specific bile acid transporters and drug metabolizing enzymes between the mouse and human components of the liver. All IHCs were conducted on the formalin fixed, wax-embedded sections of the liver and involved using antibodies to human cytokeratin 8/18, mouse cytokeratin 8/18, human albumin, human and mouse BSEP, human and mouse MRP-1, and a panel of cytochromes P450 (CYPs 3A2, 1A1, and 2B1). Table 1 gives a list of the antibody sources and the specific experimental conditions used for each antibody.
Summary of antibodies and pretreatment used in the IHC.

Note. BSEP = bile salt export pump; IHC = immunohistochemistry; MRP2 = multidrug resistance–associated protein 2; Pretreatment = antigen retrieval.
Paraffin wax, formalin fixed, blocks of liver were sectioned at 5 µm; the sections were dewaxed in xylene and brought to water before being exposed to 3% hydrogen peroxide for 10 min to block endogenous peroxidase. For the cytokeration 18, antigen retrieval was performed with EDTA buffer (pH 8.0) at 110°C for 2 min using the Milestone RHS-2 Histoprocessor (heat treatment in Table 1), with subsequent cooling for 5 min (Milestone, Shelton, CT). Alternatively sections were treated with proteinase K in 20°mM Tris-HCl, pH 8.0 (Millipore (UK) Ltd, Watford, England) at a concentration of 20 mg/ml for 10 min at room temperature. Following this pretreatment, all subsequent procedures for immunostaining were carried out in a Thermo Scientific LabVision Autostainer 720 (Lab Vision Corporation, Fremont, CA). The sites of primary antibody binding were visualized using a Dako mouse, or rabbit, HRP-labeled polymer EnVision kit (Dako UK Ltd, Ely, Cambridgeshire, UK) as appropriate for 30 min. 3,3′ diaminobenzidine hydrochloride was used as chromagen for 10 min, washed in water, and the sections were then lightly counterstained in Carazzi’s hematoxylin for 1 min and coverslipped in a Leica cv5030 automated coverslipper (Leica Microsystems, Wetzlar, Germany).
Electron Microscopy
Electron microscopy was undertaken on the samples of liver tissue that had been snap frozen in liquid nitrogen. The frozen blocks of liver were removed from the freezer and, while still frozen, slices of approximately 1 × 3 mm2 were taken from the surface of the liver, with a single-sided razor blade, and the slices were immediately plunged into 2.5% glutaraldehyde fixative in phosphate buffer of pH 7.3. The liver tissue blocks were fixed for 48 hours before being washed in phosphate buffer, postfixed in 1% osmium tetroxide in phosphate buffer for 1 hr, before being dehydrated in graded ethanols up to absolute, and then being immersed in propylene oxide, embedded in epoxy resin, and polymerized at 60°C overnight. The blocks were sectioned at 1 µm using an ultramicrotome (Leica Microsystems (UK) Ltd, Milton Keynes, England) and stained with 1% Toluidine Blue in 2% sodium borate for light microscopy examination, and selection of suitable areas for subsequent ultrathin sectioning at a thickness of approximately 100 nm. The sections were then contrasted by staining with 2% uranyl acetate in distilled water and in lead citrate (Reynolds 1963) before being examined and photographed in a Phillips CM40 electron microscope.
Results
Liver Weight
The mean body weight of the vehicle-treated SCID mouse at the start of the study was 26.8 g (range 23.4–28.9 g), while that of the vehicle-treated PXB mouse was 17.6 g (range 16.1–19.8 g). The liver to body weight ratio of the vehicle-treated PXB mouse was 13.6% as compared to a vehicle-treated SCID mouse, where the liver to body weight ratio was approximately 5.8%. Following treatment with troglitazone at 600 mg/kg, the liver to body weight ratio of the SCID mouse was 5.6% while that for the PXB mouse was 13.0% (data previously reported in Schulz-Utermoehl et al. 2012).
Morphology of the Chimeric Liver
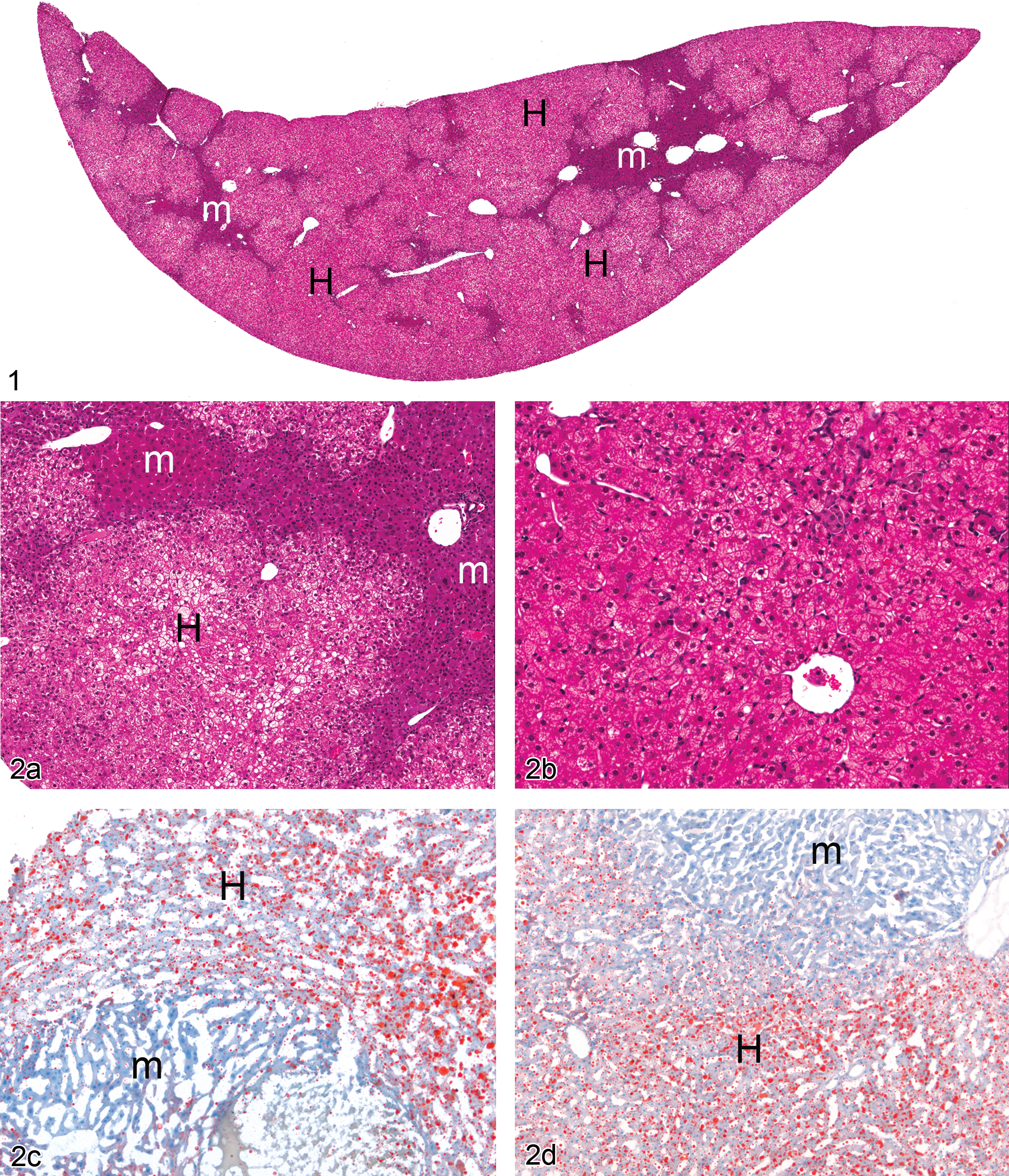
The mouse parts of the chimeric liver were present at proportions ranging from ~15% up to a maximum of ~25% of the total liver volume (Figure 1). The mean volume occupied by human hepatic portions was 79% (range 75–85%). The residual mouse parts of the chimeric liver had a strongly basophilic cytoplasm with cytoplasmic inclusions on H&E-stained sections of the PXB liver, while the human liver portions were palely eosinophilic with micro- and macrovacuolated cytoplasm (Figure 2a). The human hepatocytes were distributed in a recognizable lobular pattern with well-defined periportal and centrilobular regions. The atrophic murine portions of the PXB liver did not show any recognizable hepatic pattern in their organization although in some mice they did assume the appearance of hyperplastic foci. Treatment with troglitazone did not alter the morphology of the mouse parts of the chimeric liver but the fat vacuolation, seen in the human hepatocytes, was significantly reduced following the treatment with troglitazone (Figure 2b).
Figure 1. Vehicle-treated PXB chimeric mouse/human liver. H = human; m = murine. H&E. Figure 2. —(a) Vehicle-treated chimeric liver. Higher magnification image of junction between the human and murine parts of the liver showing the fat vacuolation in the human hepatocytes. H = human; m = murine. H&E. (b) Chimeric liver following treatment with troglitazone at 600 mg/kg for seven days showing a loss of course fat vacuolation from the human parts of the liver. H&E. (c) Vehicle-treated PXB mouse liver showing the high levels of neutral lipid in the human parts of the liver and the relatively low levels present in the murine liver. H = human; m = murine. Oil-red-O stain for lipid. (d) Troglitazone-treated PXB mouse liver showing a decrease in the amount of neutral lipid in the human parts of the liver with little change being observed in the murine liver. H = human; m = murine. Oil-red-O stain for lipid.
A summary of the Oil-red-O cytochemistry, and the IHC findings (excluding the CYP findings), in the chimeric mouse and human portions of the PXB mouse liver is shown in Table 2.
Summary of IHC results for IHC data (excluding CYPs).
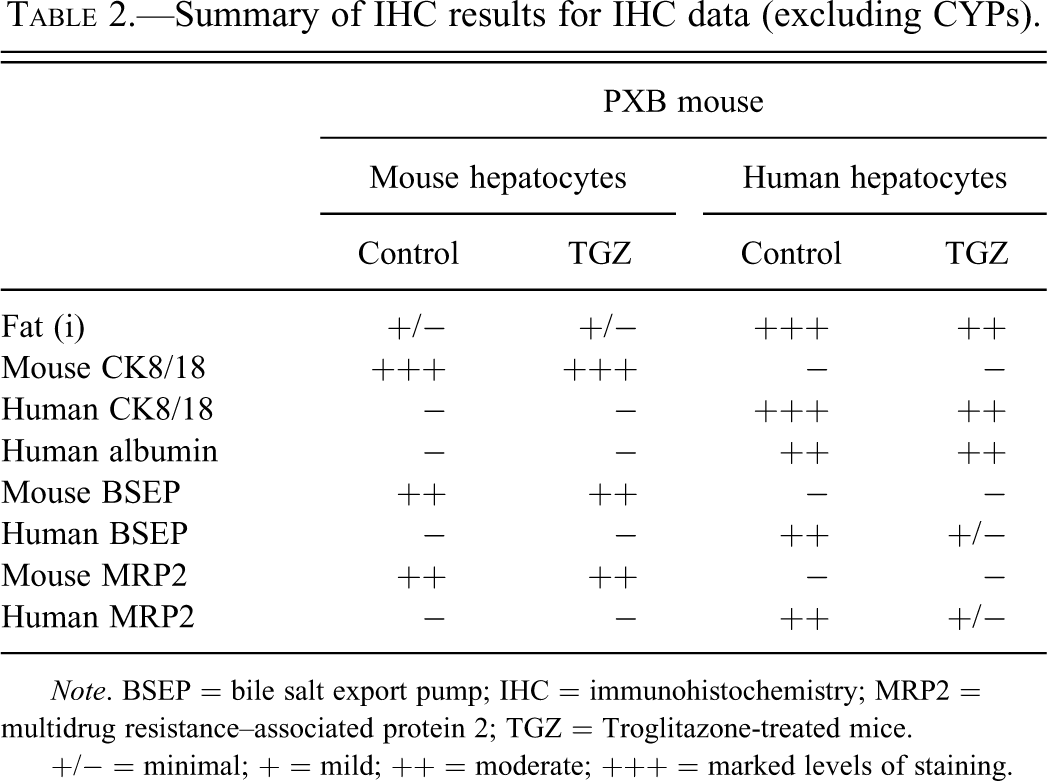
Note. BSEP = bile salt export pump; IHC = immunohistochemistry; MRP2 = multidrug resistance–associated protein 2; TGZ = Troglitazone-treated mice.
+/− = minimal; + = mild; ++ = moderate; +++ = marked levels of staining.
Oil-red-O-stained sections showed the human parts of the PXB liver to have large amounts of red-stained neutral lipid with the murine parts of the liver showing little staining for lipid (Figure 2c). Dosing with troglitazone at either 300 or 600 ppm decreased the amount of lipid present in the humanized parts of the liver while not obviously affecting the murine parts (Figure 2d).
The mouse parts of the chimeric liver stained positive with the antibody against mouse cytokeratin 8/18, showing a predominantly membranous staining; while the human parts of the chimeric liver failed to show localization of the mouse protein. When exposed to an antibody raised against human cytokeratin 8/18, the opposite staining pattern was observed with the humanized liver portions generally staining more lightly than the dense immunoreactivity shown by mouse hepatocytes stained with the antimouse cytokeratin 8/18 antibodies. The residual murine hepatocytes were generally compressed by the human parts of the liver, although the bile ducts tended to stain for both the mouse and human cytokeratin 8/18 (Figure 3a). Hepatic endothelial and Kupffer cells stained for mouse cytokeration 18 but not for the equivalent human protein (Figure 3b). Treatment with troglitazone did not change the immunostaining for cytokeratin 18 between the mouse and humanized parts of the liver.
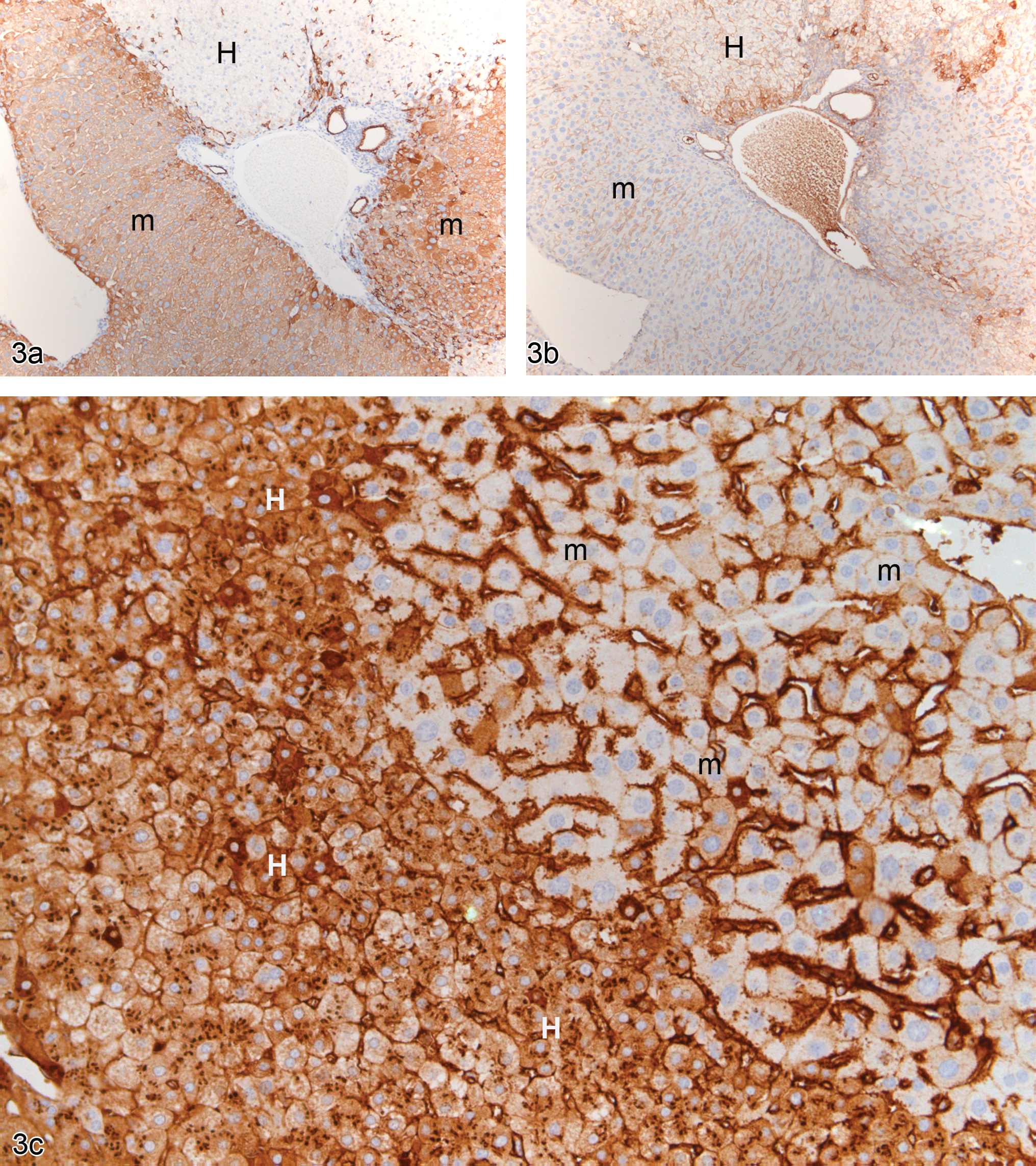
(a) Vehicle-treated PXB mouse liver stained with an antimouse cytokeratin 8/18 showing positively staining murine hepatocytes and negatively staining human hepatocytes. H = human; m = murine. (b) Vehicle-treated PXB mouse liver stained with an antihuman cytokeratin 8/18 showing positively staining human hepatocytes and negatively staining murine hepatocytes. H = human; m = murine. (c) Vehicle-treated PXB mouse liver stained with an antihuman albumin antibody showing positively staining human hepatocytes and negatively staining murine hepatocytes. The sinusoids and intracytoplasmic granules in hepatocytes in both areas of the liver are positively stained, representing the presence of human albumin. H = human; m = murine.
Sections exposed to antibodies against human albumin showed the human parts of the chimeric liver with variable cytoplasmic expression, with some cells staining strongly and others staining faintly, while the cytoplasm of the mouse hepatocytes did not show any immunostaining (Figure 3c). Protein in the sinusoids in both the human and murine hepatic areas stained intensely for the presence of human albumin.
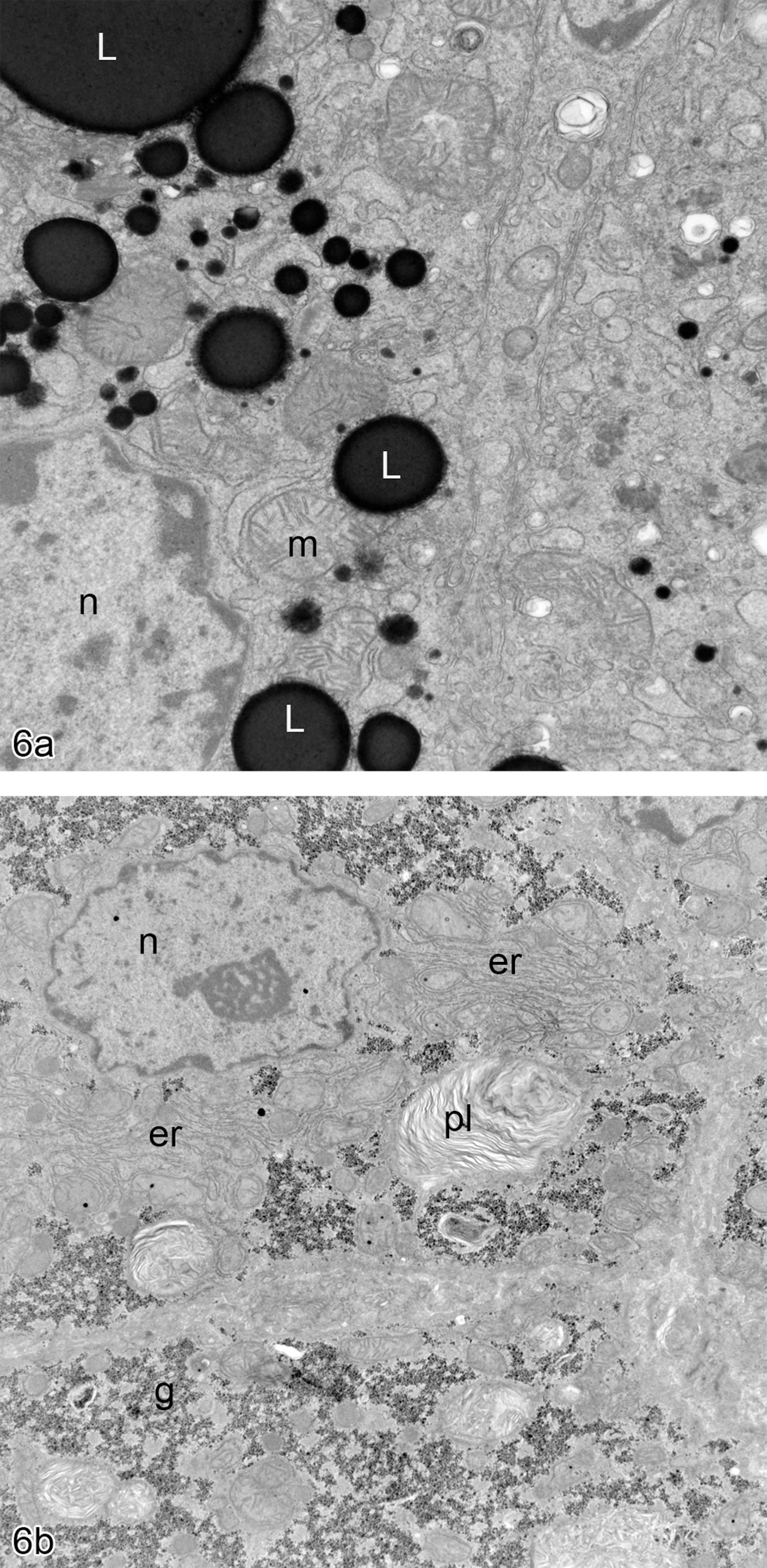
A summary table of the immunohistochemical staining for the respective CYPs is shown in Table 3. Immunostaining of the SCID mouse livers with antibodies to CYP 2B1 showed very minimal levels of expression of the apoprotein (Figure 4a), whereas in the PXB livers the residual mouse hepatocytes showed increased expression of CYP 2B1 with little if any staining being present in the human hepatocytes (Figure 4b). CYP 3A2 was highly expressed in the untreated SCID mouse liver with the majority of the hepatocytes in the centrilobular regions staining positively but also with some hepatocytes in the periportal region staining equally strongly (Figure 4c). In the PXB liver, CYP 3A2 was expressed at significantly higher levels than CYP 2B1 in the human hepatocytes, and there was a preferential centrilobular distribution (Figure 4d). As with CYP 2B1, the expression of CYP 3A2 in the residual mouse hepatocytes was considerably greater than that present in the human hepatocytes. CYP 1A1 showed an almost exclusive expression in the centrilobular hepatocytes of the SCID mouse liver, while little if any immunostaining was present in the periportal hepatocytes (Figure 4e). In contrast to the other two CYPs, CYP 1A1 was more heavily expressed in the human hepatocytes than it was in the murine hepatocytes (Figure 4F) and, as in the SCID mouse liver, the lobular distribution in the human hepatocytes of the PXB liver, favored the centrilobular areas. A small proportion of the mouse hepatocytes did stain intensely for CYP 1A1 but the majority had very low expression. It was not possible to conclude any lobular distribution for any of the CYPs in the murine hepatocyte areas, because of the lack of a defined lobular pattern shown by the residual mouse liver.
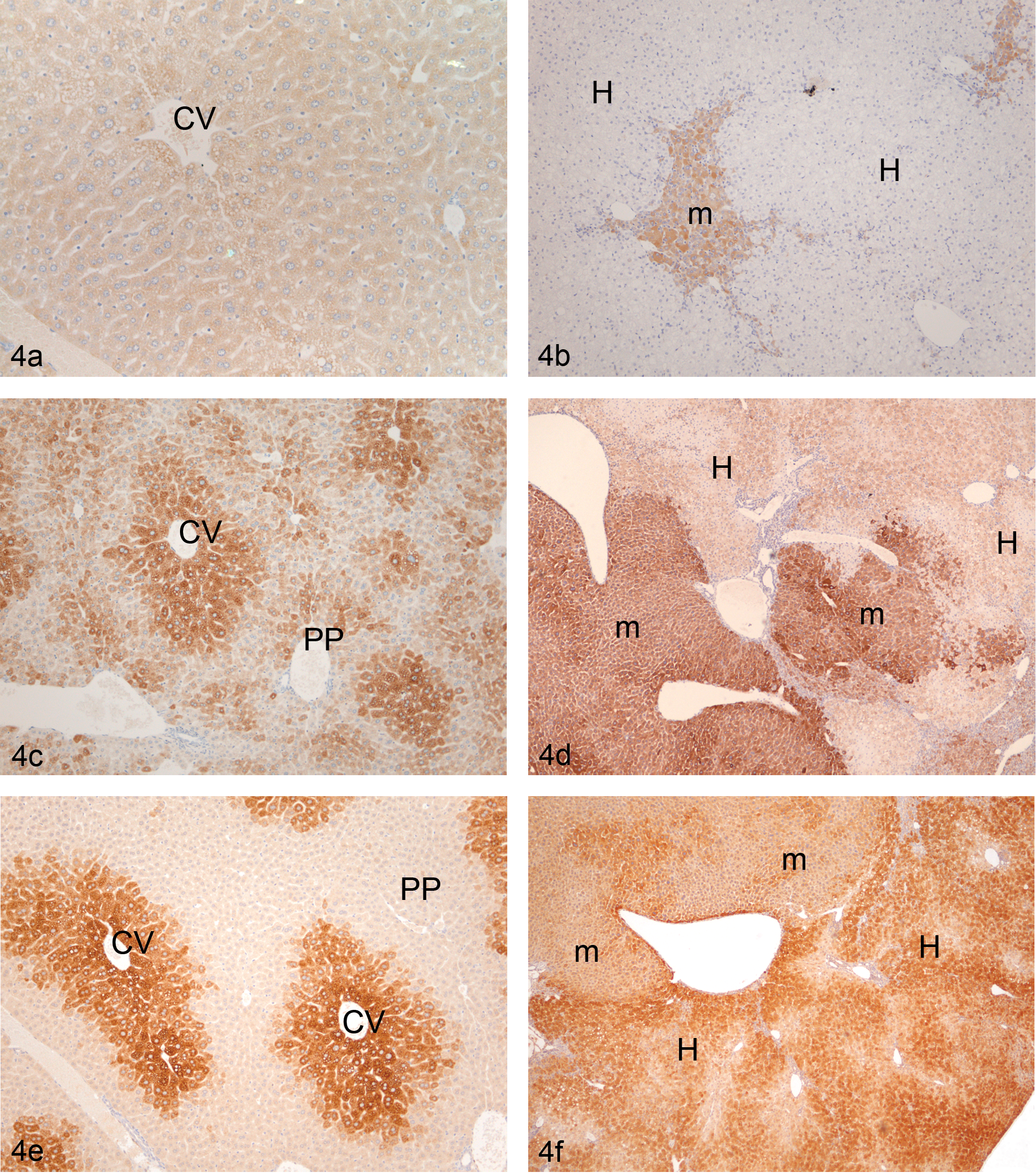
(a) Vehicle-treated SCID mouse liver stained with an anti-CYP 2B1/2 antibody showing very low expression levels present. CV = central vein. (b) Vehicle-treated PXB liver stained with an anti-CYP 2B1/2 antibody showing very low expression levels present in the human hepatocytes and the increased expression present in the residual murine liver. H = human; m = murine. (c) Vehicle-treated SCID mouse liver stained with an anti-CYP 3A2 antibody showing high constitutive expression in the centrilobular regions of the lobules (CV) with lower levels being present in the periportal regions (PP). (d) Vehicle-treated PXB liver stained with an anti-CYP 3A2 antibody showing high expression in the murine parts of the liver and lower expression levels present in the human hepatocytes, which show a centrilobular predilection for the CYP protein. H = human; m = murine. (e) Vehicle-treated SCID mouse liver stained with an anti-CYP 1A1/2 antibody showing the high constitutive expression in the centrilobular regions of the lobules (CV), with little if any staining being present in the periportal regions (PP). (f) Vehicle-treated PXB liver stained with an anti-CYP 1A1/2 antibody showing high expression in the human parts of the liver, which show a centrilobular predilection for the CYP protein, with lower expression levels, showing a less-defined distribution, being present in the murine hepatocytes, H = human; m = murine.
Summary of IHC results for cytochrome P450.
Note. IHC = immunohistochemistry; TGZ = troglitazone-treated mice.
+/− = minimal; + = mild; ++ = moderate; +++ = marked levels of staining.
Focal areas of murine hepatocytes stained more intensely.
Dosing with troglitazone did not alter the expression levels or distribution of any of the three CYPs studied, either in the SCID mouse liver or in the human or murine hepatocytes of the PXB chimeric liver (data not shown).
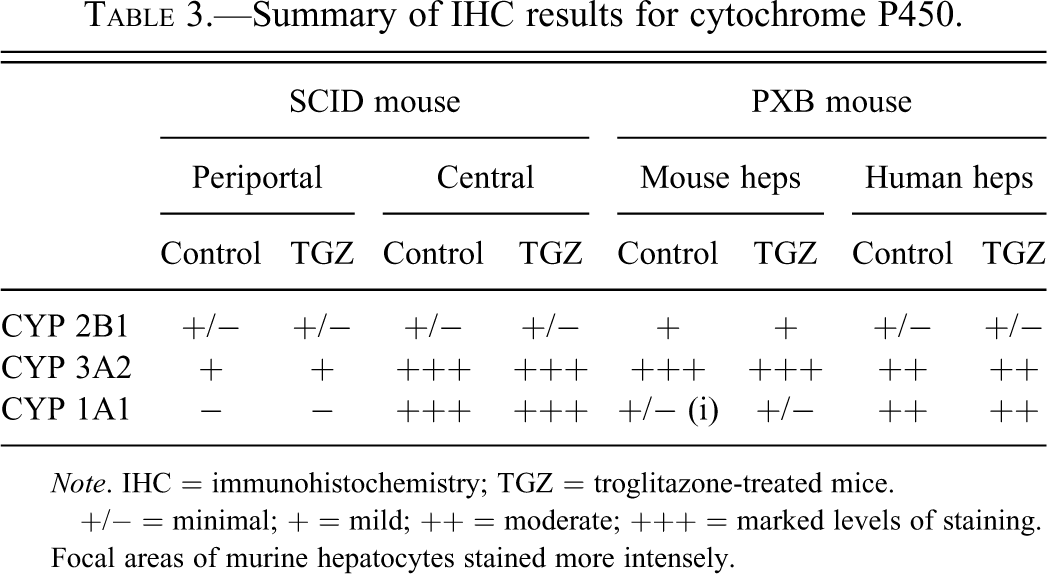
Immunostaining of the vehicle control PXB livers with antibodies to human or mouse BSEP, and human or mouse MRP2, showed the staining to be present in the regions of the canalicular membranes of the hepatocytes of both species, but generally the mouse transporters were more expressed than those in the human hepatocytes (Figure 5a and 5c). Following treatment with troglitazone, the expression of both MRP2 and BSEP remained unchanged in the mouse hepatocytes, while it was significantly decreased in the human parts of the liver (Figures 5b and 5d; data only shown for MRP2).
(a) Vehicle-treated PXB liver stained with an antihuman multidrug resistance–associated protein 2 (MRP2) antibody showing the presence of the transporter in the intercellular membranes of the human hepatocytes but no expression in the murine portions. H = human; m = murine. (b) Troglitazone-treated PXB liver stained with an antihuman MRP2 antibody showing a loss of transporter expression from the human hepatocytes. H = human; m = murine. (c) Vehicle-treated PXB liver stained with an antimouse MRP2 antibody showing the transporter in the intercellular membranes of the murine hepatocytes but not in the human hepatocytes. H = human; m = murine. (d) Troglitazone-treated PXB liver stained with an antimouse MRP2 antibody showing a similar pattern of staining to that seen in the vehicle-treated PXB mouse liver. H = human; m = murine.
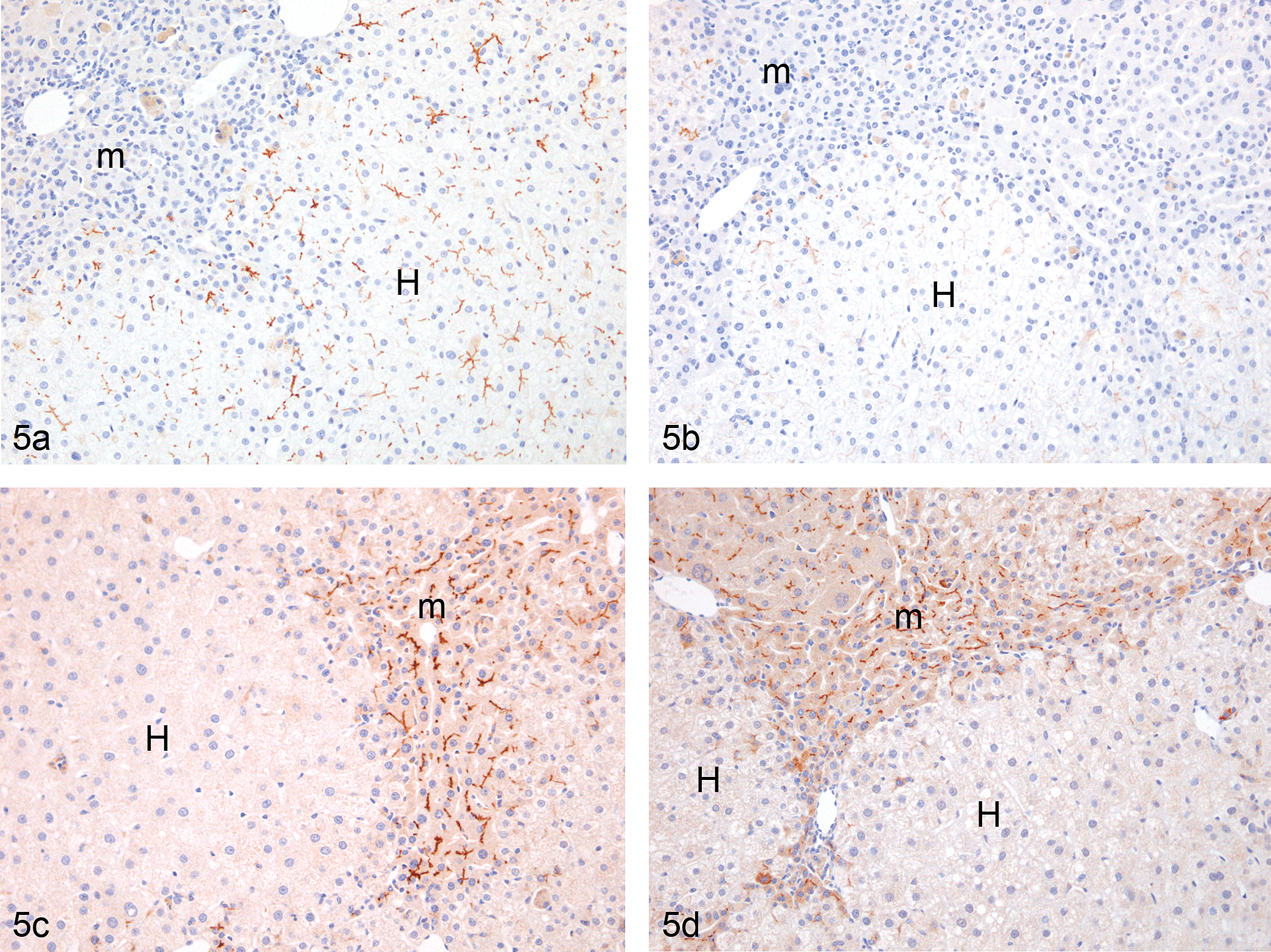
Electron microscopy of the human hepatocytes in the chimeric liver displayed an essentially normal morphology but typified by the presence of large numbers of deeply osmiophilic lipid droplets (Figure 6a) having large mitochondria and well-developed canaliculi with distinct tight junctional complexes on the membranes on each side of the canaliculi. Mouse hepatocytes showed a more rounded cellular profile with a cytoplasm containing large amounts of glycogen, increased numbers of myelinoid figures, indicative of phospholipid degradation, and moderate amounts of rough endoplasmic reticulum and mitochondria (Figure 6b). Lipid droplets were rare in the mouse hepatocytes, but wherever present they showed an electron lucid appearance with a more electron-dense border.
(a) Electron microscopy image of human hepatocyte from a vehicle-treated PXB liver showing the deeply electron-dense saturated lipid droplets (L), the nucleus (n), and mitochondria (m). (b) Electron microscopy image of murine hepatocyte from a vehicle-treated PXB liver showing the presence of phospholipid in a myelin figure (pl), the nucleus (n), glycogen (g), and rough endoplasmic reticulum (er).
Discussion
Preclinical studies of troglitazone in rodents have proven to be problematical with respect to detecting changes in relevance to the idiosyncratic hepatotoxicity seen in human patients. Significant metabolic differences between rodent and human livers are thought to be behind this failure. The current study has shown differential effects of troglitazone on the human versus a lack of detectable effects on the murine parts of the PXB chimeric liver. This was shown with regard to the downregulation of the two bile acid transporters studied, BSEP and MRP2, and a decrease in the amount of neutral lipid in the human liver. Since the drug-induced hepatotoxicity that troglitazone produces in man has a significant cholestatic component, and the two bile acid transporters in the current study are intimately concerned with bile transport, it is possible that the changes seen in the current study in the PXB mouse have direct relevance to the hepatotoxicity seen in clinical use.
The finding of lipid accumulation in the human hepatocytes within the PXB liver suggests that the environment within which they find themselves was less than optimum since hepatic lipid accumulation in vivo is taken as a sign of dysfunction (Cohen, Horton, and Hobbs 2011; French 1989; Kopec and Burns 2011; Machado and Cortez-Pinto 2011). Despite this, the other parameters assessed in the chimeric mouse liver in the current study suggest that the human cells were able to recanulate their biliary system and maintain expression of a substantially normal biochemical and functional differentiation. Indeed, published literature suggests that establishment of human cells in the PXB liver results in a return to normal of the high bilirubin levels that occur in the nontransplanted mouse (Barth et al. 2008; Meuleman et al. 2005; Sato et al. 2008), thus removing the cholestatic phenotype expressed in the failing uPA++/SCID mouse liver (Tateno et al. 2004).
The current study has shown that troglitazone differentially affects the BSEP and MRP2 bile acid transporters in the human parts of the chimeric mouse liver with no apparent effect on the residual mouse parts of the liver. Preclinical studies with troglitazone in wild-type mice showed an increase in liver weight and hepatocellular vacuolation at dose levels of 400 mg/kg in males and at dose levels of 800 mg/kg in females but overt necrosis was never seen (Herman et al. 2002). In rats, diffuse centrilobular necrosis of the liver was seen in both sexes, but this was considered to be secondary to the effects on the heart (Herman et al. 2002). In addition, hepatocellular hypertrophy was seen in the rats of both sexes (Herman et al. 2002). Hence studies in laboratory rats and mice consistently failed to show any specific hepatic effects of treatment up to dose levels that are maximally tolerated and that hold relevance to the liver damage seen in idiosyncratic liver disease in humans (Jaeschke 2007). Liver damage was observed only in SOD2+/− mice given 30 mg/kg troglitazone, which correlated with mitochondrial oxidant stress and a partial loss of mitochondrial function (Ong et al. 2007) and which was considered to better reflect the effects seen in human hepatocytes (Haskins et al. 2001) and to better predict the essential features considered to constitute toxicity in the liver in human clinical cases (Smith 2003).
The current study, while failing to show overt hepatic toxicity in the humanized PXB chimeric liver at the dose levels currently used, has shown differential effects on the human liver at the protein level, although the failure of a key bile acid transporter did not translate into a functional deficit in the form of elevations in bilirubin (data described in Schulz-Utermohl et al. 2012), which would normally be expected to rise in cases of pathological cholestasis (Berk and Javitt 1978; Fujii 1997; Handler et al. 1994). The exact reason for this may be multifactorial, including the lack of a demonstrated effect on the protein expression of another bile acid transporter, BSEP, together with other compensatory mechanisms that might operate in the mouse. Troglitazone has been shown to interfere with the biliary transport of bile acids in isolated and perfused rat livers (Preininger et al. 1999), to inhibit both BSEP and MRP2 in vitro (Funk et al. 2001a) and to inhibit bile flow in vivo in the rat (Funk et al. 2001a,b). Nevertheless, specific downregulation of the human protein to MRP2 in the chimeric liver is evidence of differential metabolism of troglitazone in the human versus the mouse hepatocytes, as is shown in life (He et al 2001; Honma et al. 2001; Yamazaki et al. 1999; Yamamoto et al. 2002), and is supportive of the maintenance of a humanized metabolism in the PXB chimeric liver that is clearly absent from the mouse hepatocytes.
The expression of two of the three CYP drug metabolizing enzymes currently studied, CYPs 2B1 and 3A2, showed higher expression in the residual mouse liver than that in the human parts of the chimeric liver. In contrast, the expression levels of CYP 1A1 were higher in the human parts of the chimeric liver than that in the murine parts. Expression of all three CYPs was unchanged following treatment with troglitazone at either 300 or 600 ppm, in either the mouse or human liver parts. CYP 3A has been shown to be important in the generation of reactive metabolites of troglitazone (Yokoi 2010), and the differential expression of the enzyme between the mouse and the humanized liver might have predicted an increased production by the mouse liver. This apparent discrepancy may be explained by the differences between the expression of the apoprotein, displayed by the antibody to the CYP, and the actual enzymic activity. This question would need to be addressed by the separation of the mouse and the human liver compartments in subsequent metabolism studies. It would also be interesting to speculate as to whether the hepatic levels of CYPs in the murine parts of the chimeric liver were higher than those present in the uPA++/SCID mouse liver before transplantation of the human hepatocytes. It is clear that the expression of CYP 2B1 in the residual mouse hepatocytes in the chimeric liver is induced over that present in untreated mouse liver, where expression levels of this enzyme are normally extremely low (Warner, Fernstrom, and Ruch 2003).
In a series of studies to determine whether the chimeric livers were responding metabolically in a human, rather than a murine way, chimeric mice have been given a small range of chemicals/drugs and their response to chemical exposure assessed. Rifampicin and the polycyclic aromatic hydrocarbon, 3-methylcholanthrene, specific inducers of hepatic CYP 3A and CYP 1A1 and 1A2, respectively, when given by intraperitoneal injection induced the mRNA levels of six CYPs when the livers were assessed 24 hours later (Bowen et al. 2000). Rifampicin increased the mRNA of CYP 3A4 approximately 6-fold, while 3MC administration increased the mRNA levels of CYP 1A1 by 10-fold and those of CYP1A2 by 6.4-fold. A follow-up study with acetaminophen administered to the uPA++/SCID chimeric mice also showed a differential response with the human portions of the chimeric liver being considerably less sensitive to acetaminophen toxicity than were wild-type ICR mice given the same dose level (Sato et al. 2008). Once again, this suggested that the livers of the chimeric mice were responding metabolically in a more humanized, rather than a murine, way.
In conclusion, the study on the effects of troglitazone on the PXB mouse liver has demonstrated differential effects on the hepatocytes of the two species, with downregulation of bile acid transporters being shown by the human hepatocytes that was not evident in the mouse equivalent. Concomitant drug metabolism studies, performed on samples taken from this study, provided metabolic profiles that were similar to those reported in man and from which it was concluded that the PXB mouse model could provide a useful first insight into circulating human metabolites of xenobiotics metabolized in the liver (Schulz-Utermoehl et al. 2012). From the data presented here, it would appear that this system, the PXB mouse liver, has the potential to allow in vivo studies of human-specific metabolism of potentially toxic drugs that could never be ethically administered to man, while in addition permitting histopathological investigations on necropsied mice at the end of the dosing periods. Its potential use in drug development for mechanistic and drug metabolism/disposition studies is enormous, once the complete biological characterization of the model system has been achieved.
Footnotes
Acknowledgments
The authors acknowledge the help of Mr. Kevin Randall and Mr. Richard Jenkins for preparing the IHC and Mr. Simon Brockelhurst for preparing and photographing the electron microscopy.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.