
Abstract
Inflammatory myofibroblastic tumors (IMTs), originating from mesenchymal cells, are rare neoplasms with intermediate biological potential. Despite being predominantly benign, they can progress to locally aggressive disease and may recur over time. IMTs are predominantly seen in the pediatric population, making our case of IMT in an older adult even rarer. A 67-year-old female with a 42-pack-year smoking history presented with cough, fever, and progressively worsening exertional dyspnea. Imaging studies, including chest X-ray followed by chest computed tomography, identified a right lower lobe lung nodule without evidence of metastasis. Subsequent evaluation with robotic bronchoscopy and endobronchial ultrasound revealed densely cellular reactive lung tissue on lymph node biopsy. Ultimately, the patient underwent a robotic-assisted right lower lobectomy. Final pathology confirmed the diagnosis of an IMT. IMTs are characterized by the presence of spindle cells associated with dense monomorphic inflammatory cells. It has been found that IMT arises from chromosomal rearrangements that aberrantly activate various kinase signaling pathways. This understanding of the molecular mechanisms underlying IMT development has elucidated the neoplastic nature of the disease and has also been pivotal in distinguishing IMT from other inflammatory pseudotumors. Surgical resection remains the cornerstone of treatment, with targeted therapies offering promising results for unresectable or advanced cases. IMTs pose a multifaceted challenge in clinical practice due to their diverse clinical manifestations, histopathological variability, and unclear etiopathogenesis, underscoring the need for continued research and a multidisciplinary approach to optimize patient outcomes.
Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare mesenchymal neoplasm characterized by proliferation of myofibroblastic and fibroblastic spindle cells accompanied by a prominent inflammatory infiltrate composed of plasma cells, lymphocytes, eosinophils, and histiocytes. 1 Historically considered a reactive inflammatory process, advances in molecular pathology have established IMT as a true neoplasm. Approximately 50% to 80% of cases harbor chromosomal rearrangements involving the anaplastic lymphoma kinase (ALK) gene locus, with alternative kinase fusions involving ROS1, NTRK, RET, and PDGFRB identified in ALK-negative cases.2,3 IMT most commonly occurs in the abdominal cavity, retroperitoneum, and lung, particularly in children and young adults, with a median age of 11 years in large pediatric series.4,5 Pulmonary IMT constitutes <1% of all adult lung tumors. 6
Although IMT is more common in younger patients, it can occur across all age groups. Recent studies have documented cases in elderly adults, with some series reporting mean ages of 47 to 54 years in pulmonary IMT.7-9 Importantly, recent evidence challenges the notion that ALK expression decreases with age, demonstrating ALK rearrangements in 91% of IMTs in patients ≥40 years. 8 Diagnosis requires histopathologic evaluation, as fine-needle aspiration and bronchoscopic biopsy have suboptimal diagnostic yield. 6 Complete surgical resection remains the mainstay of treatment for localized IMT, with excellent long-term outcomes.9,10 Large multiinstitutional studies have demonstrated an overall 5-year survival of 95% and a 5-year event-free survival of 80%.4,5 For unresectable, recurrent, or metastatic disease, ALK inhibitors such as crizotinib have demonstrated remarkable efficacy in ALK-rearranged IMT, with response rates exceeding 90% in some series.2,3
Despite the well-characterized predilection for younger patients, IMT occurring in elderly adults remains underreported in the literature. We present an unusual case of pulmonary IMT in an elderly female patient who underwent successful surgical resection and is currently under observation.
Case Description
A 67-year-old female presented with nasal congestion, cough, nausea, and fever associated with exertional shortness of breath a few weeks before the initial presentation. She has a 42-pack-year smoking history, a remote history of basal cell cancer of the nose, and a family history of lung cancer in her mother. Vital signs were stable. She had adventitious breath sounds on the exam. Chest X-ray revealed a right lower lobe nodule. Computed tomography (CT) of the chest showed a right lower lobe lung nodule measuring 1.6 × 1.5 × 1.3 cm without evidence of metastasis. Positron emission tomography showed intensely fluorodeoxyglucose (FDG) avid anteromedial right lower lobe nodule measuring 1.4 cm (Standardized uptake value (SUV) 11.6) with mild FDG in the right superior hilum (Figure 1).
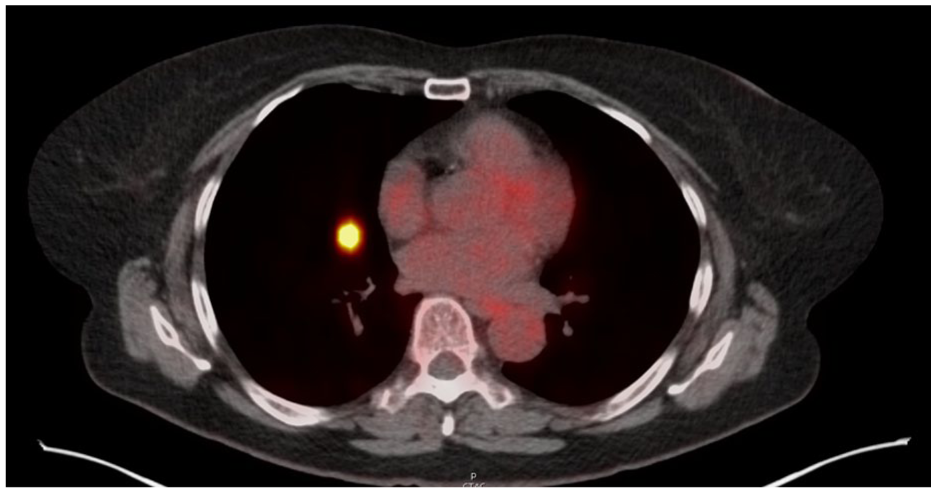
Axial PET/CT chest shows intensely FDG avid anteromedial right lower lobe nodule measuring 1.4 cm (SUV 11.6) with mild FDG in the right superior hilum. CT, computed tomography; FDG, fluorodeoxyglucose.
The pulmonary function test showed a decreased Diffusion capacity of the lungs for carbon monoxide (DLCO) without an obstructive or restrictive pattern. The echocardiogram showed an ejection fraction of 55% to 65% with normal systolic and diastolic function. She subsequently underwent a robotic bronchoscopy with endobronchial ultrasound. The pathology of the right lower lobe nodule revealed densely cellular reactive lung tissue with reactive type-2 pneumocyte hyperplasia and interstitial expansion, characterized by spindle cells, lymphocytes, and plasma cells (Figure 2a–c).

Histopathologic features of IMT. (a) Low-power view (H&E, ×40) showing a well-circumscribed spindle cell lesion. (b) Medium-power view (H&E, ×100) highlighting fascicular arrangement and inflammatory infiltrate. (c) High-power view (H&E, ×400) demonstrating spindle cells with vesicular nuclei and eosinophilic cytoplasm. (d) Immunohistochemistry showing diffuse cytoplasmic positivity for ALK-1 in tumor cells. ALK-1, anaplastic lymphoma kinase-1; IMT, inflammatory myofibroblastic tumor.
The diagnosis was not definitive, but pathology discouraged a repeat biopsy because it is unlikely to resolve the diagnostic dilemma. Consequently, she underwent robotic-assisted right lower lobe lobectomy and wedge resection of the middle lobe along with mediastinal lymph node dissection. Pathology revealed an IMT with negative margins and without lymphovascular invasion or visceral pleural involvement. ALK-1 was positive (Figure 2d). The lymph nodes were negative for malignancy. Patient’s next generation sequencing resulted in TPM4-ALK rearrangement, TP53 missense variant with variant allele fraction (VAF) of 2.3%, and DNMT3A frameshift mutation with VAF of 2.3%.
The case was discussed in a multidisciplinary tumor board, and the recommendation was surveillance with CTs and consideration for systemic therapy if recurrence occurs. During follow-up, she reported intermittent dyspnea on exertion but has remained stable without recurrence for 2 years now.
Discussion
IMT is an uncommon mesenchymal neoplasm characterized histologically by the abnormal proliferation of spindle-shaped myofibroblasts accompanied by a chronic inflammatory infiltrate. 11 Initially thought to be reactive lesions and referred to as inflammatory pseudo-tumors, IMTs have since been reclassified as true neoplasms of intermediate biological potential. 12 The exact etiology remains uncertain, but the most widely accepted hypothesis suggests that IMTs arise from an aberrant inflammatory or postinfectious response. 11 This shift in classification is primarily due to accumulating evidence, including the identification of ALK gene rearrangements, which supports their neoplastic nature. 13 Although generally benign, IMTs have the potential for local recurrence and, in rare cases, distant metastasis. 12
IMTs most commonly affect children and young adults, with the lung being the most frequent site of involvement. Pulmonary IMTs account for <1% of all primary lung tumors. 14 Clinical presentations are often nonspecific, characterized by symptoms such as cough, dyspnea, chest pain, or hemoptysis. A significant proportion of cases are discovered incidentally during imaging studies performed for unrelated reasons. 12 Radiologically, pulmonary IMTs typically present as solitary, well-circumscribed masses, often located in the peripheral lung fields. While CT scans are typically the preferred imaging modality, specific characteristics on MR diffusion-weighted imaging and apparent diffusion coefficient maps have been documented that aid in distinguishing IMT from infectious processes and lung cancer. 15 While imaging may suggest a benign or low-grade lesion, it is not diagnostic, as IMTs can mimic malignant tumors, infectious granulomas, and other spindle cell neoplasms. 12 Transthoracic fine-needle aspiration biopsy and bronchoscopic specimens are usually too small and insufficient for a definite diagnosis. 16 Therefore, surgical excision of the lesion is the preferred method of diagnosis. 17 Following this, histopathological confirmation remains essential. 13
Histologically, IMTs are composed of spindle-shaped myofibroblasts set in a collagenous stroma infiltrated by plasma cells, lymphocytes, and eosinophils. 18 Immunohistochemically, the spindle cells typically express smooth muscle actin and desmin, and ALK positivity further supports the diagnosis. 11 Recent molecular studies have revealed recurrent chromosomal rearrangements, particularly involving the ALK gene. 13 ALK reengagements on 2p23 account for close to 50% of IMTs.11,19,20
ALK becomes oncogenic through fusion with other genes, most commonly tropomyosin genes (TPM3 or TPM4) in IMT. Other fusion partner genes identified in IMT include RANBP2, TFG, CARS, ATIC, LMNA, PRKAR1a, CLTC, FN1, SEC 31A, and EML4. 21 EML4-ALK was initially described only in lung cancer; however, now, after a few studies, they constitute up to 20% of ALK-rearranged IMTs. Most of these are noted in pulmonary IMTs. 22 The diagnosis of ALK-negative IMTs is often tricky due to a markedly variable phenotype. ALK-negative IMTs may be more aggressive than ALK-positive tumors, with a higher frequency of metastasis and limited treatment options for unresectable/advanced disease. 21 Most ALK-negative tumors harbor gene fusions affecting the tyrosine kinase pathway, including ROS1 rearrangements at 6q22, NTRK, RET, and PDGFRβ mutations.21,23 ROS1 gene fusions include TFG and YWHAE. 24
The most comprehensive recent study by Bakhshwin et al specifically examined IMTs in adults ≥40 years and found that 91% of cases showed ALK expression through immunohistochemistry and/or molecular testing. 8 This directly contradicts earlier reports suggesting ALK positivity decreases with age. The study explicitly states: “Despite earlier reports, ALK is frequently expressed/rearranged in tumors in this age group.” 8 However, there is a clear age-related difference in the overall frequency of kinase fusions (not just ALK). Antonescu et al demonstrated that while 56% of all IMTs showed ALK rearrangements, >90% of pediatric IMTs showed gene rearrangements (including ALK, ROS1, and other kinases), whereas 90% of fusion-negative IMTs were seen in adults. 22 This suggests that while ALK-positive IMTs can occur in older adults at high rates, fusion-negative IMTs are more common in the adult population overall.
Conventional detection methods such as Immunohistochemistry (IHC)and Fluorescence in situ hybridization (FISH) may miss a range of actionable gene rearrangements, underscoring the need for broader molecular testing with Next Generation Sequencing (NGS). In adult IMTs, ~50% to 70% harbor kinase fusions when standard testing is used, but this increases to 70% to 85% with comprehensive molecular profiling. 25 The remaining 15% to 30% of adult IMTs are fusion-negative, representing a significantly higher proportion than in pediatric populations, where fusion-negative cases are rare (<10%). These findings have diagnostic and therapeutic implications. 23 The presence of TP53 missense mutation (VAF 2.3%) and DNMT3A frameshift mutation (VAF 2.3%) in this case is noteworthy, as TP53 mutations are uncommon in fusion-positive IMTs and typically associated with more aggressive behavior in ALK-negative cases.26,27 The low VAFs suggest these may represent clonal hematopoiesis or minor sub clonal populations rather than dominant driver mutations. 27
Complete surgical excision remains the gold standard of treatment and is typically curative, with most patients experiencing favorable outcomes and low recurrence rates. 14 In instances where surgery is not ideal due to either multifocality, location, disease spread, or associated patient comorbidities, alternatives such as chemotherapy and radiation are available. Multiple classes of chemotherapy drugs, including platinum-based chemo, taxanes, methotrexate, and etoposide, have been used. A high dose of radiation therapy, 40 to 50 Gy, is usually required, with some case reports of successful treatment with low doses. 28 The role of steroids in treatment remains controversial, with limited evidence of benefit and occasional reports of disease progression. 17 An exception may be IgG4-related IMTs, which have shown some response to steroid therapy.
In cases where surgery is not feasible due to anatomical location, comorbidities, or recurrence, targeted therapy with ALK inhibitors has shown promise, particularly in ALK-positive tumors. 12 The advent of targeted therapy with the TK inhibitor crizotinib, as demonstrated in trials such as the “EORTC trial 90101 CREATE,” has indeed shown promising results for advanced or unresectable tumors. Crizotinib is an inhibitor of ALK, MET, and ROS1. 23 Neoadjuvant therapy with ALK inhibitors is being used for locally advanced tumors, but data are limited. 19 Additionally, alectinib, ceritinib, and lorlatinib are newer-generation tyrosine kinase inhibitors (TKIs), each offering enhanced efficacy and safety profiles compared to older agents, such as Crizotinib. Entrectinib is a pan-TRK, ROS1, or ALK inhibitor. Sequential treatment for ALK-TKI is being explored to prolong survival. 29 While the overall behavior of IMT is indolent, rare instances of aggressive behavior or metastasis have been reported, emphasizing the need for long-term clinical follow-up. 13
Conclusion
IMTs present a multifaceted challenge in clinical practice due to their diverse clinical manifestations, histopathological variability, and unclear etiopathogenesis. Despite its rarity, advancements in molecular understanding and diagnostic imaging have enhanced our ability to differentiate IMT from other inflammatory processes and malignancies. Surgical resection remains the cornerstone of treatment, with targeted therapies, such as crizotinib and newer-generation TKIs, offering promising results for unresectable or advanced cases. However, challenges persist in accurately diagnosing and managing IMT, emphasizing the importance of continued research efforts and a multidisciplinary approach to optimize patient outcomes.
Footnotes
Author Note
Prior Presentation of Abstract Statement: This case was previously presented as only as an abstract “Inflammatory Myofibroblast Tumor of Lung: A Rare Find in an Elderly Female” at CHEST 2024, October 06 to 09, 2024 in Boston Convention and Exhibition Center, Boston, Massachusetts, USA.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent to participate
Written informed consent was obtained from the patient for their anonymized information to be published in this article.
Consent for Publication
Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.