
Abstract
As severe conditions, acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) threaten human health. Inflammation and oxidative stress play a vital role in the pathogenesis of ALI/ARDS. Sphingosine kinase 1 (SphK1) significantly contributes to mediating inflammatory responses. Nevertheless, the impact of SphK1 on lipopolysaccharide (LPS)-triggered ALI/ARDS remains largely undetermined. In our current work, we explored the impact of SphK1 on ALI/ARDS using a mouse model. We studied whether it could reduce LPS-triggered inflammatory response and oxidative stress by suppressing SphK1 in ALI/ARDS. The mice were treated with the inhibitor of SphK1 (N,N-dimethylsphingosine, DMS) before intraperitoneal injection of LPS. Moreover, we assessed the survival rate, and several parameters, such as the lung wet/dry (W/D) ratio, myeloperoxidase (MPO) activity, superoxide dismutase (SOD) activity, malondialdehyde (MDA) content, and the release of inflammatory cytokines. Western blotting analysis was adopted to evaluate the levels of phosphoinositide 3-kinase (PI3K)/serine/threonine kinase (AKT) pathways. We showed that the inhibitor of SphK1 not only ameliorated LPS-stimulated lung histopathological changes and W/D ratio of lung tissue but also elevated the survival rate, the SOD activity and decreased the MDA content, MPO activity, interleukin-6 (IL-6) and tumor necrosis factor-ɑ (TNF-ɑ) production by regulating the PI3K/AKT signaling pathway in lung tissue. Taken together, SphK1 played an essential role in inflammatory responses and oxidative stress. The underlying mechanism might be linked to the activation and up-regulation of the PI3K/AKT signaling pathway in LPS-triggered ALI/ARDS.
Keywords
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) dramatically contribute to morbidity and mortality today.1,2 These two conditions are characterized by overwhelming inflammation, oxidative stress, alveolar barrier dysfunction, and immune cell infiltration. Oxidative stress and inflammation are inseparably associated with ALI/ARDS.3,4 In the experimental animal model of ALI/ARDS, lipopolysaccharide (LPS) can trigger systemic inflammation to release pro-inflammatory mediators,5,6 such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), leading to increased accumulation of reactive oxygen species (ROS). 7 In addition, superoxide dismutase (SOD) activity and malondialdehyde (MDA) content are related to LPS-induced oxidative stress. 8 Previous reports have revealed that inhibiting inflammation and oxidative stress can mitigate LPS-triggered ALI/ARDS. 3 Therefore, finding effective inhibitors against inflammatory response and oxidative stress is crucial.
Several investigations have reported that ALI/ARDS can induce an inflammatory reaction and oxidative stress by activating the phosphoinositide 3-kinase (PI3K)/serine/threonine kinase (AKT) signaling.9,10 Glycyrrhizic acid inhibits the release of inflammatory cytokines in LPS-triggered ALI/ARDS by modulating autophagy mediated by the PI3K/AKT/mTOR pathway. 11 Furthermore, the inflammatory and antioxidant effects of 3-O-trans-caffeoyloleanolic acid are regulated by its impacts on PI3K/AKT pathway. Therefore, inhibiting PI3K/AKT pathway is considered a practical approach for treating many inflammatory diseases. 12
As a crucial rate-rater, sphingosine kinase 1 (SphK1) functions as an enzyme responsible for transducing intracellular signals in sphingolipid metabolism.13,14 Up-regulation of SphK1 can increase the proliferation, angiogenesis, migration, invasion, metastasis, inflammation, and metabolism of many tumor cells.15–17 After activating SphK1, pro-inflammatory factors, such as ROS and Toll-like receptor (TLR) signals, and various pro-inflammatory pathways are activated.18,19 In addition, the expression level of SphK1 remains high in several types of immune cells and carcinomatous cells,20–22 and it promotes inflammation by activating pro-inflammatory cytokines. 23 Nevertheless, the specific impacts of SphK1 on ALI/ARDS remain largely unexplored. It has been demonstrated that up-regulation of SphK1 can trigger the PI3K/AKT/NF-κB signaling, 24 and blocking the PI3K/AKT/NF-κB signaling abolishes the anti-apoptotic properties of SphK1 on non-small-cell lung cancer (NSCLC) cells.25,26 Transfection of miRNA-103 can reduce the expression of SphK1, induce apoptosis, inhibit cell proliferation, and down-regulate the PI3K/AKT signaling activated by SphK1 to promote osteoarthritis (OA) treatment. 27 Therefore, inhibition of SphK1 can reduce the PI3K/AKT signaling activity, 28 leading to reduced expressions of pro-inflammatory mediators. 29 However, there are few reports on whether inhibiting SphK1 can inactivate the PI3K/AKT signaling to ameliorate LPS-triggered ALI/ARDS.
N,N-dimethylsphingosine (DMS), an inhibitor of SphK1, can inhibit cell proliferation and induce apoptosis in human lung cancer cell line A549. 30 It can down-regulate the release of inflammatory factors, such as galectin-3, interferon-γ, and TNF-α, and cardiac inflammatory mediators in serum of Chagas disease cardiomyopathy, thereby reducing the inflammatory response. 31
According to previous studies, it remains largely unclear whether SphK1 facilitates inflammation and oxidative stress via the PI3K/AKT signaling in LPS-triggered ALI/ARDS. Therefore, we planned to explore the impacts of SphK1 on inflammation and oxidative stress on LPS-triggered ALI/ARDS with DMS using a mouse model. We proposed that the activation of PI3K/AKT was the critical regulatory mechanism associated with the impacts of SphK1.
Materials and methods
Materials
N,N-dimethylsphingosine (DMS) was provided by Sigma-Aldrich (St. Louis, MO). LPS was supplied by Sigma Chemical Co. (L-2880, St. Louis, MO, USA). The MPO and MDA assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Enzyme-linked immunosorbent assay (ELISA) kits for mouse IL-6 and TNF-α were obtained from BioLegend (CA, USA). All primary antibodies were supplied by Abcam (Cambridge, MA, USA), and a horseradish peroxidase (HRP)-conjugated secondary antibody was provided by Proteintech (Chicago, IL, USA). Other reagents were of analytical grade.
Animal grouping and modeling
C57BL/6 mice (male, 6-8 weeks old) were obtained from Hunan Slake Jingda Experimental Animals Co., Ltd. (Changsha, China). All animals were bred in a climate-controlled facility (25 ± 2°C) with a relative humidity of 55 ± 10% and a 12-h light/dark photoperiod. All animal-related protocols were authorized by the Medical Ethics Committee of Jishou University (No.2020030), and experimental procedures complied with the Chinese Council on Animal Care guidelines. Our research was conducted at Jishou University from October 2020 to December 2021. A total of 80 BALB/c mice were randomly assigned into four groups: control group, control + DMS group, ALI group, and ALI + DMS group. Mice were intraperitoneally injected with DMS [10 μM in phosphate-buffered saline (PBS)]or PBS before the LPS challenge. 32 The ALI/ARDS model was established by 10 mg/kg LPS (Sigma-Aldrich, St. Louis, MO) via intratracheal instillation or sham-treated with normal saline (NS). Bronchoalveolar lavage fluid (BALF), serum samples, and lung tissue were harvested 24 h after the LPS challenge.
Collection of samples
Collection of serum
At 24 h after LPS induction, the mice were subjected to anesthesia using 3% pentobarbital, and the abdomen was opened to expose the trachea and lungs. In addition, blood was harvested from the heart. After the whole blood was incubated at 37°C for 2 h, the upper serum was centrifuged at 3500 rpm for 15 min at 4°C and preserved at −80°C.
Collection of BALF
Briefly, 0.8 mL PBS was slowly given by intratracheal instillation thrice, followed by immediate suction and rinsing, and then BALF was spun at 1500 rpm for 10 min at 4°C. Next, the supernatant was collected and subjected to the protein content assay using the bicinchoninic acid (BCA) kit.
Collection of lung tissue
After collection of BALF, the lung tissue was removed and fixed in 1 mL of precooled 4% paraformaldehyde overnight. Part of the lung tissue was used for the tissue section. The other part of the lung tissue was rinsed with aseptic saline, the excess water was removed by filter paper, and the sample was frozen at −80°C.
Histopathological evaluation of lung tissue
The lung tissue was harvested at 24 h after the LPS exposure, followed by fixation in 4% paraformaldehyde. Then, the fixed lung tissue was dehydrated with an ethanol gradient. Next, paraffin-embedded tissues were sliced into 5-μm sections, dewaxed with xylene, and rehydrated with an ethanol gradient. According to the instructions of the reagent, the sections of each group were stained using a hematoxylin and eosin (H&E) solution. Subsequently, the sections were visualized under a microscope (Leica Microscope Ltd., Wetzlar, Germany) at a magnification of ×10 to assess the morphological changes in the lung. According to a modified scoring system by Hasan et al., five random areas were graded on a scale from 0 to 4 (0, absent and appears normal; 1, light; 2, moderate; 3, strong; 4, intense). This scoring system included size of alveolar spaces, thickness of alveolar septa, alveolar fibrin deposition and neutrophil infiltration, and the median value was calculated in each tissue sample. The histological score yielded a maximum total score of 16. 33
MPO assay
Myeloperoxidase activity in the lung was analyzed by its detection kit. Briefly, lung tissues were subjected to homogenization. Then, 0.9 mL of tissue homogenate was mixed with 0.1 mL buffer, followed by incubation for 15 min. The MPO activity was determined at a wavelength of 460 nm using a Multiskan JX microplate reader (Thermo Labsystems, ThermoBioAnalysis, Tokyo, Japan).
Wet to dry (W/D) ratio of the lung
The wet weight of each group was obtained using a fresh right lung. Subsequently, the lungs were dried at 80°C for 48 h to get the dry weight. According to these weights, the W/D ratio of the lung was determined.
Determination of TNF-α and IL-6 contents
IL-6 and TNF-α in the serum and BALF were assessed using ELISA kits at 450 nm.
Determination of SOD activity and MDA content
To assess the antioxidant defenses, the enzyme activities in the BALF and lung tissues were determined. In addition, the contents of SOD and MDA in BALF and lung tissue homogenates were examined. The lung tissue was homogenized and spun at 3000-4000 rpm for 10 min. Subsequently, the supernatant was harvested to prepare 10% tissue homogenate, and the SOD activity and MDA content were measured using the detection kits. The supernatant and BALF were adopted on the same day to assay the oxidative biomarkers, MDA and SOD.
Western blotting analysis
The lung tissue (100 mg) was lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitor and phosphatase inhibitor and incubated on ice for 30 min, and the total protein of the lung tissue was extracted. The total protein content was quantified by the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Briefly, equal amounts of total proteins (about 30–40 μg) were subjected to SDS-PAGE and then transferred onto PVDF membranes. The membranes were blocked in a blocking buffer at room temperature for 1 h or at 4°C overnight. The blots were incubated with primary antibodies against PI3K P85 (rabbit, 1:1,000, ab191606, Abcam), p-PI3K P85 (phospho Y607) (rabbit, 1:500, ab182651, Abcam), AKT (rabbit, 1:10,000, ab179463, Abcam), p-AKT (phospho S473) (rabbit, 1:10,000, ab81283, Abcam), and GAPDH (rabbit, 1:10,000, ab181602, Abcam) overnight on a shaker at 4°C. The blots were rinsed with TBST thrice (5 min each). Next, the blots were incubated with an HRP-conjugated secondary antibody (goat anti-rabbit IgG, 1:2,000, SA00001-2, Proteintech) at room temperature for 60 min. Immunoreactive bands were visualized using an ECL chemiluminescence detection kit. GAPDH was adopted as the loading control. The band intensity was quantified with ImageJ software.
Statistical analyses
All experiments were performed at least three times (n ≥ 3), and the experimental variables were expressed as mean ± SEM. One-way ANOVA was adopted to analyze the differences among groups. Bonferroni’s multiple comparisons were employed as post-test analyses. Survival data were analyzed. Histology scores were employed as Kruskal-Wallis analyses. A p-value <0.05 was considered statistically significant.
Results
Blockade of SphK1 attenuates the lung damage and increases the survival rate in mice with ALI/ARDS
In the current work, we explored the impact of Sphk1 on the pathological changes in lung tissue induced by LPS. Figure 1(a) shows that the typical structure of lung tissue was found in the control group. In contrast, the lung tissue in the ALI group showed significant pathological changes, such as neutrophil infiltration, edema, thickened alveolar wall, and alveolar disorder. DMS treatment inhibited pathological changes in lung tissues induced by the LPS challenge (Figure 1(a)). Likewise, histology scores were higher in the ALI group compared with the control group, while scores declined in the ALI + DMS group (Table 1). These findings showed that SphK1 could pathologically damage the lungs in ALI/ARDS and deteriorate the infiltration of inflammatory cells. Collectively, these data indicated that suppression of SphK1 could mitigate the pathogenesis and progression of ALI/ARDS. Impacts of Sphk1 on pathological lung tissue changes in LPS-triggered ALI/ARDS. (a) Lung tissues; (H&E staining, magnification ×10; n = 5). (b) Survival rate. (*p < .05 vs. control, #p < .05 vs. ALI, n = 20). Lung histology scores. *p < .05, n = 5.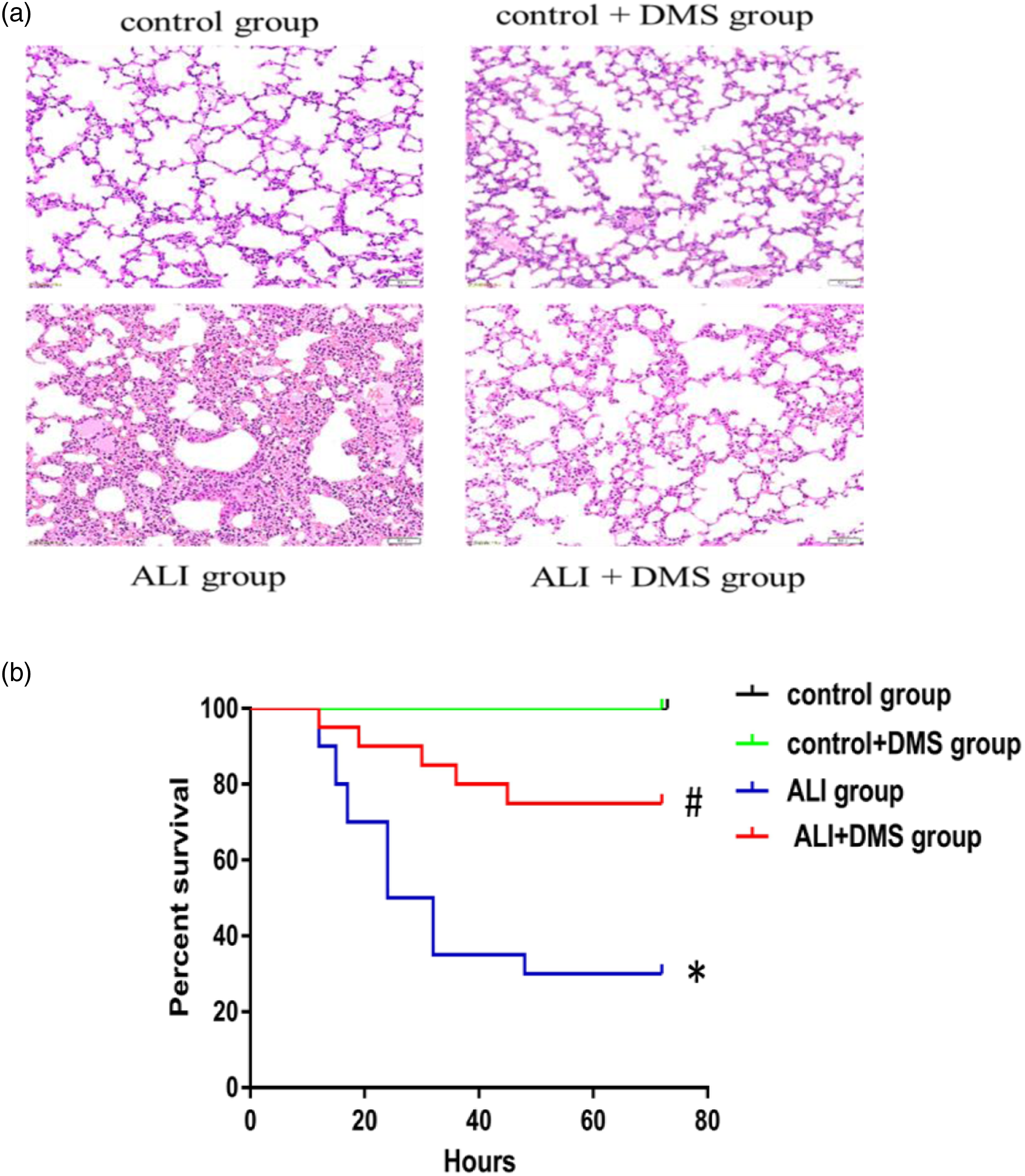

To assess the effect of SphK1, we explored the survival rate during ALI/ARDS. ALI/ARDS mice in the control group and control + DMS group survived. In the ALI group, the mortality was 50% at 24 h post-injection. Kaplan-Meier analysis revealed a difference in survival between the groups (p < .05). The result suggested that SphK1 could reduce mortality during ALI/ARDS (Figure 1(b)).
SphK1 increases the lung W/D ratio and MPO activity in ALI/ARDS
LPS-triggered ALI/ARDS is characterized by pulmonary edema. We determined the lung W/D ratio to assess pulmonary edema in the present work. We found that the LPS challenge remarkably enhanced the pulmonary W/D ratio (p < .05). Nevertheless, DMS decreased such W/D ratio (p < .05) (Figure 2(a)). In addition, MPO, a marker of neutrophil infiltration, was examined. LPS remarkably enhanced MPO activity in lung tissue (p < .05). However, DMS significantly reduced LPS-induced MPO activity (p < .05) (Figure 2(b)). These findings implied that SphK1 played a vital role in the inflammatory reaction in ALI/ARDS. Impacts of SphK1 on pulmonary edema and inflammatory response in ALI/ARDS. It was performed to detect the lung W/D ratio and MPO activity. Values are expressed as mean ± SEM (*p < .05, vs. control; #p < .05, vs. ALI, n = 10).
SphK1 decreases the activity of SOD and increases the level of MDA in ALI/ARDS
The MDA content and SOD activity in the lung tissues and BALF were examined to examine the impacts of SphK1 on changes in oxidant-antioxidant systems. As an antioxidant enzyme, SOD can scavenge ROS to achieve antioxidant capacity. On the other hand, MDA is a marker of oxidative damage. Figures 3(a) and (c) show that the SOD activity in the ALI group was markedly decreased compared with the control group (p < .05). However, the SOD activity was still lower than the control group even after DMS treatment, while it was remarkably elevated than that of the ALI group (p < .05). The MDA content in the ALI group was enhanced (p < .05). However, such value in the ALI + DMS group was dramatically reduced than that of the ALI group (p < .05) (Figures 3(b) and (d)). The data indicated that SphK1 could reduce the SOD activity and increase the MDA content in ALI/ARDS. Taken together, SphK1 played an essential role in enhancing oxidative stress in ALI/ARDS. Effects of SphK1on oxidative stress in ALI/ARDS. SOD and MDA in both BALF and lung tissue homogenate were determined. DMS decreased the SOD activity and increased the MDA level in lung tissues, and the same result was obtained in BALF. Data are expressed as mean ± SEM (*p < .05, vs. control; #p < .05, vs. ALI, n = 10).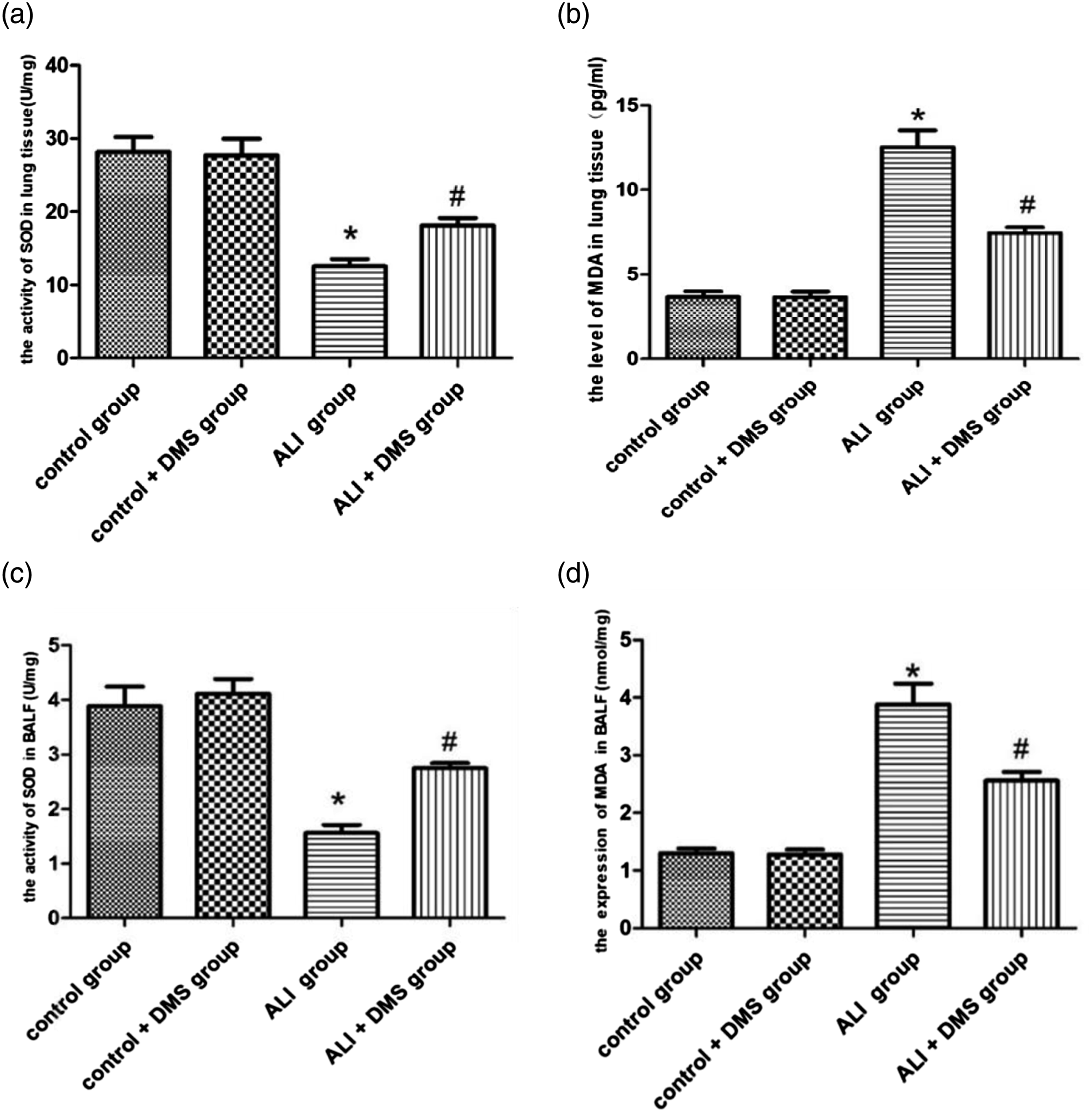
SphK1 enhances the contents of TNF-α and IL-6 in serum and BALF of mice with ALI/ARDS
In order to determine whether SphK1 influenced inflammation of LPS-triggered ALI/ARDS, we analyzed the production of TNF-α and IL-6, which are the two most critical pro-inflammatory cytokines secreted by macrophages. Their excessive secretion can result in extensive inflammation and tissue damage, playing a vital role in lung injury. These data implied that in serum and BALF, the levels of TNF-α and IL-6 were elevated in the ALI group than those of the control group (p < .05), while those values in the ALI + DMS group were evidently decreased than those of the ALI group (p < .05) (Figure 4(a)–(d)). These findings implied that inhibition of SphK1 effectively weakened the levels of TNF-α and IL-6 in ALI/ARDS, indicating SphK1 played a fundamental role in accelerating inflammation in ALI/ARDS. Impacts of SphK1 on inflammation in ALI/ARDS. (a) The level of TNF-α in serum; (b) The level of IL-6 in serum; (n = 10); (c) The level of TNF-α in BALF; (d) The level of IL-6 in BALF. Data are expressed as mean ± SEM (*p < .05, vs. control; #p < .05, vs. ALI, n = 10).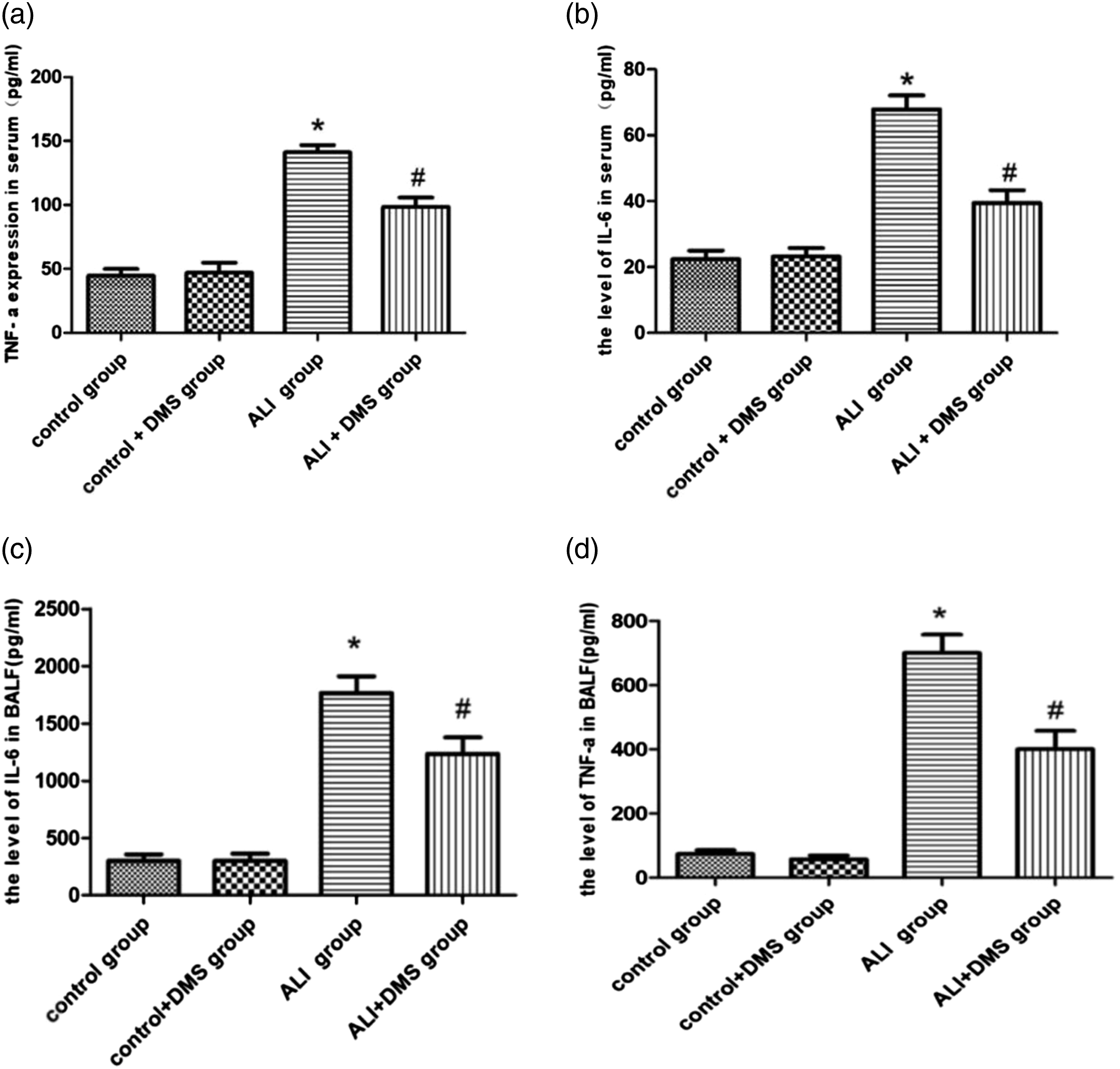
SphK1 facilitates the deposition of PI3K/AKT in lung tissues of mice with ALI/ARDS
The PI3K/AKT signaling pathway plays a vital role in the inflammatory response and oxidative stress. The present work adopted Western blotting analysis to assess the PI3K/AKT deposition in the lung tissues. We found that the contents of PI3K, p-PI3K, AKT, and p-AKT proteins were enhanced in the ALI group than those of the control group. However, compared with the ALI group, these proteins were down-regulated in the ALI + DMS group. These results showed that inhibition of SphK1 was related to down-regulated activation of the PI3K/AKT signaling pathway (Figure 5). Therefore, SphK1 significantly contributed to inflammation and oxidative stress. The underlying mechanism might be linked to the activation and up-regulation of the PI3K/AKT signaling pathway in LPS-triggered ALI/ARDS. PI3K/AKT protein expression. (a) Representative Western blotting of PI3K/AKT and (b) the results of the quantitative densitometric analysis. (*p < .05,**p < .01,****p < .0001, n = 3).
Discussion
In our current work, mice with ALI/ARDS were administered with the inhibitor of SphK1. These findings revealed that SphK1 contributed to inflammation and oxidative stress by impacting the activation of MPO, SOD, MDA, TNF-α, and IL-6 in ALI/ARDS. Besides, we found that SphK1 aggravated inflammation and oxidative stress by up-regulating the PI3K/AKT signaling. Therefore, SphK1 was a therapeutic intervention target for ALI/ARDS.
To explore the impact of SphK1, we established an ALI/ARDS mouse model by intratracheal instillation of LPS. DMS was used as an inhibitor of SphK1 in this study. It reduces the release of TNF-α, IL-1β, and IL-6 in the serum of mice with LPS-induced acute liver failure (ALF), thereby enhancing the survival rate of mice. 34 DMS can effectively inhibit the expression of SphK1 and alleviate the occurrence and development of many diseases.
In this study, suppression of SphK1 decreased pathologic destruction and increased survival. Suppression of SphK1 remarkably attenuated the infiltration of immune cells in the alveolar and interstitial spaces of lung tissue, and the lung W/D ratio was also reduced. It suggested that SphK1 contributed to organ damage and the survival rate during ALI/ARDS.
As a sensitive and particular marker for ALI/ARDS, 35 the MPO assay was used to detect the activity of neutrophils in tissues. As expected, we found that suppression of SphK1 decreased the MPO activity in lung tissues compared with the ALI group, suggesting that SphK1 could promote immune cell infiltration in the lungs after the LPS exposure. TNF-α enhances the permeability of pulmonary epithelium and further triggers lung injury, leading to neutrophil infiltration and lung edema in ALI/ARDS. 36 IL-6 is a critical biomarker in monitoring ALI/ARDS. 37 Up-regulation of pro-inflammatory cytokines, such as TNF-α and IL-6, contributes to inflammation. 38 Our findings revealed that SphK1 could facilitate systemic inflammation in ALI/ARDS by up-regulating TNF-α and IL-6.
Oxidative stress damage plays a vital role in LPS-triggered ALI/ARDS. 39 Oxidative stress results in the overproduction of ROS, MPO, and MDA and reduces the expression of SOD, promoting lung tissues from oxidative damage in ALI/ARDS. 40 In our experiments, the blockade of SphK1 elevated the SOD activity and reduced the MDA content in LPS-triggered ALI/ARDS. These findings suggested that SphK1 could enhance inflammation and oxidative stress in LPS-triggered ALI/ARDS.
According to the above-mentioned findings, we further clarified the mechanism underlying the impact of SphK1 on LPS-caused inflammation and oxidative stress in ALI/ARDS. Previous investigations have revealed that ALI/ARDSPI3K/AKT signaling pathway contributes to inflammation and oxidative stress.41–43 In addition, previous reports have also shown that suppressing the PI3K/AKT signaling pathway mitigates LPS-triggered ALI/ARDS. 44 As a specific inhibitor of the PI3K/AKT signaling, LY294002 can markedly inhibit LPS-triggered NF-κB activation and decrease TNF-α and IL-6.45,46 Furthermore, the PI3K/AKT signaling regulates oxidative stress in pulmonary inflammation.47,48 miRNA-103 plays a fundamental role in chondrocyte apoptosis, which can facilitate OA progression by down-regulating the PI3K/AKT signaling by reducing SphK1 activity. 27 Atractylenolide-1 is a potent candidate for treating colitis by inhibiting inflammation via the SPHK1/PI3K/AKT axis. 24 In conclusion, SphK1 could mediate the occurrence and development of many diseases by regulating PI3K/AKT. However, the association between SphK1 and PI3K/AKT in LPS-induced ALI/ARDS is largely unknown, and the underlying mechanism is poorly understood. In the present work, the blockade of SphK1 down-regulated the activation of the PI3K/AKT pathway in LPS-caused ALI/ARDS. Possibly, SphK1 might mediate LPS-triggered ALI/ARDS by modulating the PI3K/AKT signaling pathway.
However, in our study there are some limitations. Firstly, our results were obtained from ALI animal models experiments. Results from cell experiments are lacking. Second, the present study only demonstrates a small part of inflammatory cytokines and oxidative stress index. Finally, this study needs to be future confirmed by clinical studies. Further studies are warranted.
Conclusions
Taken together, SphK1 played an essential function in the inflammatory response and oxidative stress in LPS-triggered ALI/ARDS. Moreover, SphK1 promoted lung damage and increased inflammatory response and oxidative stress in ALI/ARDS by regulating the PI3K/AKT signaling.
Footnotes
Acknowledgements
The authors would like to thank Hunan Provincial Department of Education for their support.
Author contributions
Xiao-Li Wang designed the trial. Chao-shun Wei and Xiao-Li Wanghave conducted the work and are involved in data collection. . Chao-shun Wei analyzed the data and interpreted the data. Chao-shun Wei and Xiao-Li Wang wrote the manuscript. All authors revised the manuscript and contributed significantly to this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Hunan Provincial Department of Education Research Project (No. 18A286).
Ethics approval and consent to participate
The study was obtained permission from the Biomedical Ethics Committee of Jishou University (JSDX-2021-0022).
Animal welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.