
Abstract
Kikuchi-Fujimoto disease, also known as histiocytic necrotizing lymphadenitis, is characterized by high fever, lymph node swelling, and leukopenia. It is usually a benign self-limited disease. However, there are reports that it can be associated with other conditions, including infectious and noninfectious processes, autoimmune diseases such as systemic lupus erythematosus (SLE), or even life-threatening conditions like macrophage activation syndrome (MAS). Here, we report 2 cases of Kikuchi disease with non-self-limited disease in West Texas. The first case involves a 13-year-old Hispanic female who presented with prolonged fever for 8 weeks, cervical lymphadenopathy, and malaise. A year later, she was diagnosed with SLE. The second case is a 36-year-old Hispanic female who presented with prolonged fever and cervical, supraclavicular, axillary lymphadenopathy, and pancytopenia. She then developed MAS or hemophagocytic lymphohistiocytosis.
Keywords
Introduction
Kikuchi-Fujimoto disease (KFD), also called histiocytic necrotizing lymphadenitis, is a relatively rare and usually self-limited condition, which is characterized by subacute necrotizing regional lymphadenopathy. The disease was first described in Asia by Kikuchi and Fujimoto in 1972 and later in the United States and Europe. Although initially believed to primarily affect Asians, KFD is now recognized across various ethnic groups, though its exact prevalence remains poorly studied. 1 Young adults under 40 years old are typically affected but can range from 6 and 80 years old. The incidence of KFD is higher in females than in males, with a ratio of approximately 1.5:1 to 4:1. Patients commonly present with cervical lymphadenopathy, fever, malaise, and weight loss, which can lead to misdiagnosis as infection, lymphoma, or metastatic tumor. The diagnosis of KFD is based exclusively on lymph node histopathology and mostly made retrospectively. Hallmark findings of FKD consist of extracellular debris and intracellular apoptotic debris embedded in the cytoplasm of crescentic and phagocytic.1-3 Here, we present 2 cases of Hispanic patients with Kikuchi disease who experienced consequences or complications: one with systemic lupus erythematosus (SLE) and another with macrophage activation syndrome (MAS).
Case 1 Description
A 13-year-old Hispanic, previously healthy female, presented with prolonged fever for 8 weeks. She had lost 15 pounds in the previous 4 months. She denied any recent travel, sick contacts, or exposure to pets or cats. There was no prior history of recurrent infections or hospitalizations, and no family history of autoimmune disease or cancer. She developed a prolonged fever with upper respiratory tract symptoms and was treated empirically with a 10-day course of cefdinir. Despite the treatment, her fever persisted along with abdominal pain. She was admitted to a hospital and underwent extensive infectious and rheumatologic workup, which was unremarkable. An ultrasound of the abdomen revealed cholelithiasis, leading to cholecystectomy. During that admission, she was anemic and leukopenic. Bone marrow evaluation revealed hypocellular marrow with negative cytogenetic myelodysplastic syndrome. A computed tomography (CT) scan revealed numerous enlarged cervical lymph nodes, more notable on the left side, with central necrosis, and in the supraclavicular area. A CT scan of the chest, abdomen, and pelvis did not reveal enlarged lymph nodes. A left cervical lymph node biopsy revealed follicular paracortical hyperplasia with foci of necrotizing lymphadenitis. The infectious workup from the lymph node biopsy was negative. She received empiric intravenous immunoglobulins (IVIG) for 2 doses with slight clinical improvement. The patient was discharged with further outpatient investigation.
However, she was subsequently admitted to another hospital due to worsening anemia requiring blood transfusion, leukopenia, and a low-grade fever. A repeat bone marrow aspirate and biopsy showed similar results. A repeat cervical lymph node biopsy revealed necrotizing lymphadenitis. Additional outside reports of the first lymph node biopsy revealed paracortical and sinusoidal expansion with increased histiocytes and mixed infiltrates. The myeloperoxidase stain was negative with no Hodgkin cells or Epstein-Barr virus (EBV) cells seen. The histopathological diagnosis suggested KFD. Immunoglobulin levels were normal. Blood and urine cultures, cytomegalovirus (CMV) IgM, EBV IgM, Bartonella serology, toxoplasma serology, coccidioidomycosis serology, and QuantiFERON-TB Gold (QFT) were all negative. Screening tests for autoimmune diseases included antinuclear antibody (ANA), anti-double-stranded DNA antibodies (anti-dsDNA), rheumatoid factor (RF), and antineutrophil cytoplasmic antibodies (ANCA) negative. A diagnosis of Kikuchi syndrome was, therefore, established by exclusion. She was treated with steroids with the resolution of her symptoms; she was symptom-free for a year.
She presented 1 year later with subacute fever, lymphadenopathy, and generalized discoid lupus erythematosus as Figure 1. Laboratory tests were significant for mild anemia, leukopenia with low-normal platelets, and low complement levels, with positive ANA, dsDNA antibody, and Smith antibody, fulfilling ACR/EULAR 2019 criteria for SLE. She was admitted and treated with 2 doses of 80 mg IV methylprednisolone and 300 mg daily hydroxychloroquine (HCQ). The patient was discharged on prednisone 40 mg daily, hydroxychloroquine, clobetasol solution, and ketoconazole shampoo. Due to difficulty in controlling the disease, mycophenolate mofetil (MMF) and weekly subcutaneous Belimumab (200 mg) were added during follow-up. Her condition remained well controlled; however, shortly after tapering her corticosteroids, she developed intermittent abdominal pain and chronic watery diarrhea, leading to hospitalization due to hypotension. Upon admission, she was found to have anemia, leukopenia, transaminitis, hyperferritinemia, elevated inflammatory markers, and low complement levels, as shown in Table 1. Abdominal CT angiography revealed no evidence of vasculitis. Esophagogastroduodenoscopy (EGD) showed chronic gastritis, while colonoscopy demonstrated chronic colitis histologically. No medication adjustments were made at that time. She was treated with high-dose methylprednisolone, resulting in symptom improvement. During follow-up with her rheumatologist, Ruxolitinib was introduced for disease control and as a steroid-sparing agent. She has been doing well and on tapering prednisone, hydroxychloroquine, Belimumab, and Ruxolitinib with controlled SLE.
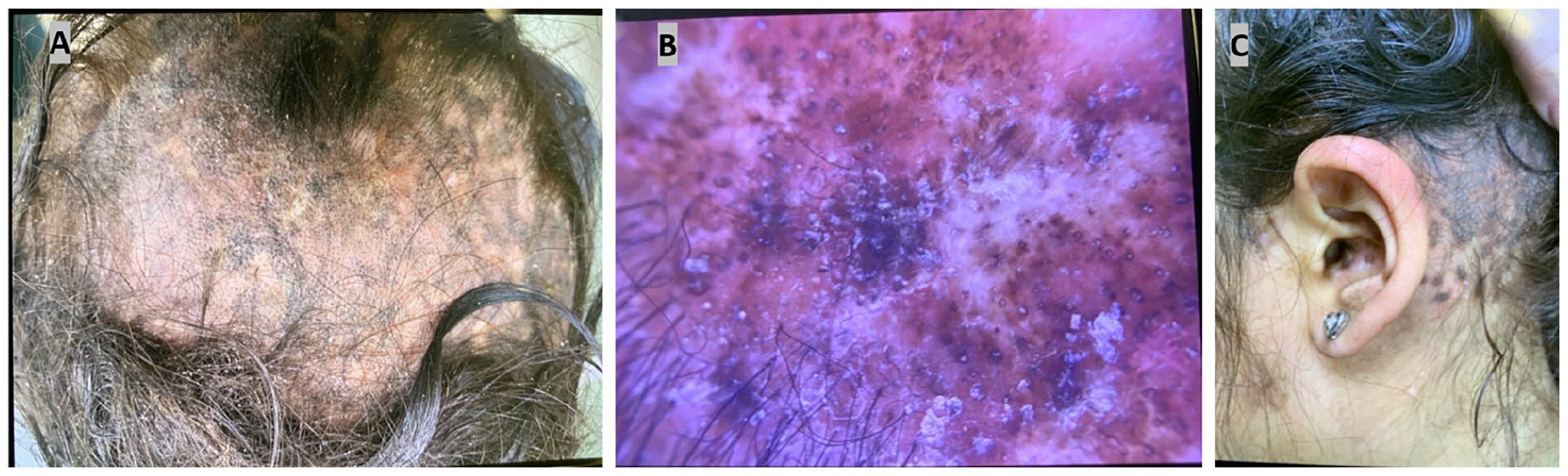
Physical examination revealed scaly, erythematous-violaceous, hyperpigmented patches, and plaques diffusely distributed on the frontal scalp (A, B), along with cicatricial alopecia and bilateral conchal bowl involvement (C).
Summarized Laboratory Investigation of Case 1.
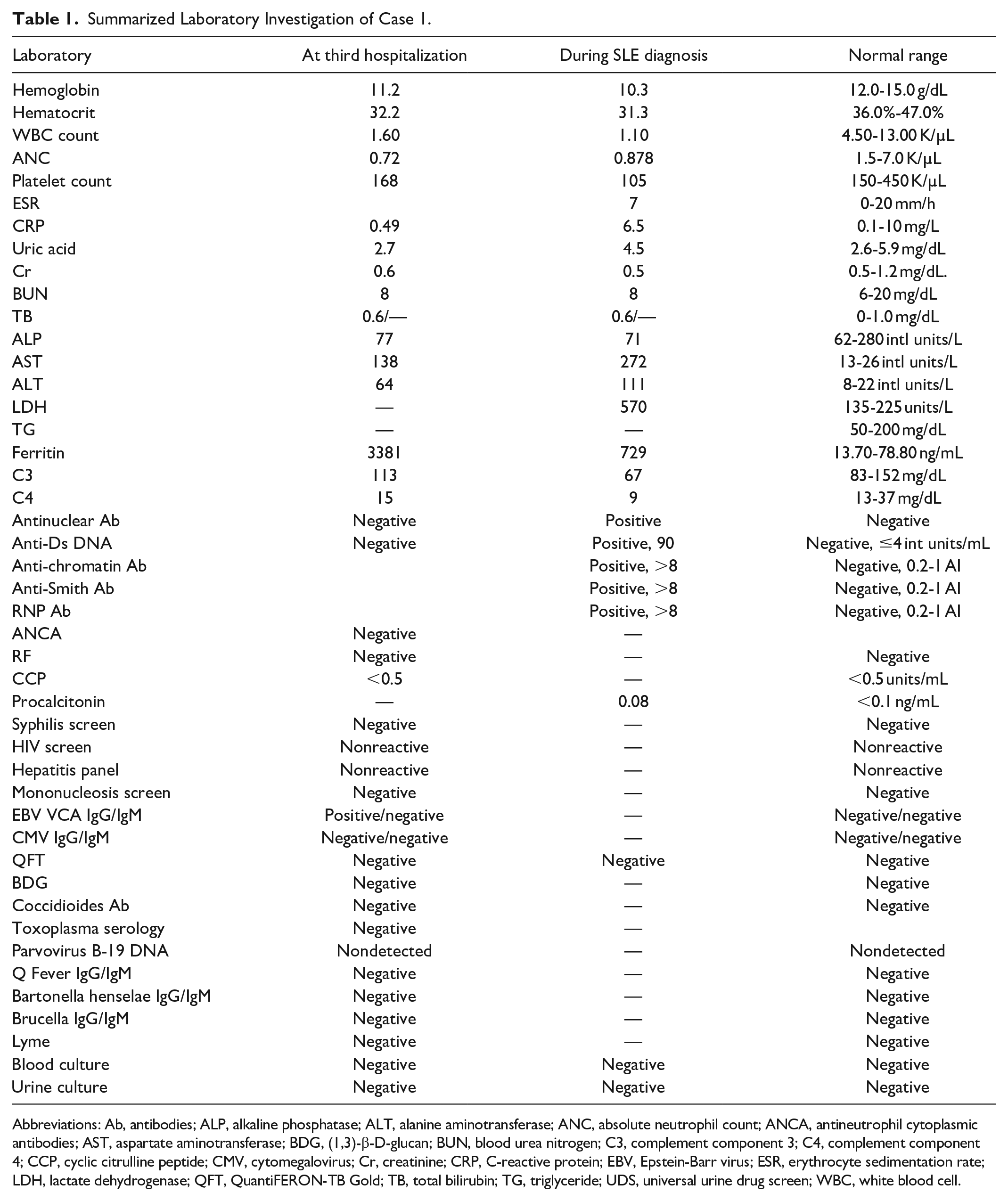
Abbreviations: Ab, antibodies; ALP, alkaline phosphatase; ALT, alanine aminotransferase; ANC, absolute neutrophil count; ANCA, antineutrophil cytoplasmic antibodies; AST, aspartate aminotransferase; BDG, (1,3)-β-D-glucan; BUN, blood urea nitrogen; C3, complement component 3; C4, complement component 4; CCP, cyclic citrulline peptide; CMV, cytomegalovirus; Cr, creatinine; CRP, C-reactive protein; EBV, Epstein-Barr virus; ESR, erythrocyte sedimentation rate; LDH, lactate dehydrogenase; QFT, QuantiFERON-TB Gold; TB, total bilirubin; TG, triglyceride; UDS, universal urine drug screen; WBC, white blood cell.
Case 2 Description
A 36-year-old Hispanic woman with no significant medical history presented to the hospital with a 3-week fever occurring daily, typically twice a day in the morning and evening. Associated symptoms included back pain and headache. She denied nausea, vomiting, cough, shortness of breath, dysuria, pain, night sweats, or rashes. She had been managing her symptoms with acetaminophen and ibuprofen, which provided temporary relief. The patient reported no recentsick contact, travel outside of West Texas, exposure to medications, or contact with livestock or pets. A course of cephalexin was completed without improvement.
Upon physical examination, the patient was noted to have a maximum temperature of 101.5°F and cervical lymphadenopathy. Initial laboratory investigations revealed leukopenia, anemia, low-normal platelet count, mild elevation aspartate aminotransferase/alanine aminotransferase, elevated C-reactive protein and erythrocyte sedimentation rate, and lactate dehydrogenase levels of 747 units/L. Additional laboratory tests are shown in Table 2. Transthoracic echocardiography did not reveal any vegetation. A CT scan of the chest, abdomen, and pelvis showed small para-aortic lymph nodes with mild haziness. A neck CT scan without contrast revealed diffuse lymphadenopathy in the neck and upper chest. A peripheral blood smear showed pancytopenia. A bone marrow biopsy demonstrated normocellular bone marrow with dysplastic features and normal cytogenetics. Cytogenetic and Fluorescence In Situ Hybridization (FISH) studies were normal without features of hemophagocytosis. A core-needle lymph biopsy of the cervical lymph node showed necrotic lymphadenopathy. Immunohistochemical (IHC) stains of CD3, CD20, CD79a, and CD45 highlighted reactive staining patterns; IHC stains of CD30, and acid-fast bacillus were negative. A left axillary lymph node excision was repeated revealing extensive necrosis and marked apoptotic debris consistent with Kikuchi disease, without features of lymphoma, other malignancy, or definite infectious etiology shown in Figure 2. The lymphocytes are mostly small without significant cytologic atypia. A few viable areas with intact lymphoid cells are seen. Additional laboratory investigations for atypical and fungal infections were unremarkable, as seen in Table 2.
Summarized Laboratory Investigation of Case 2.
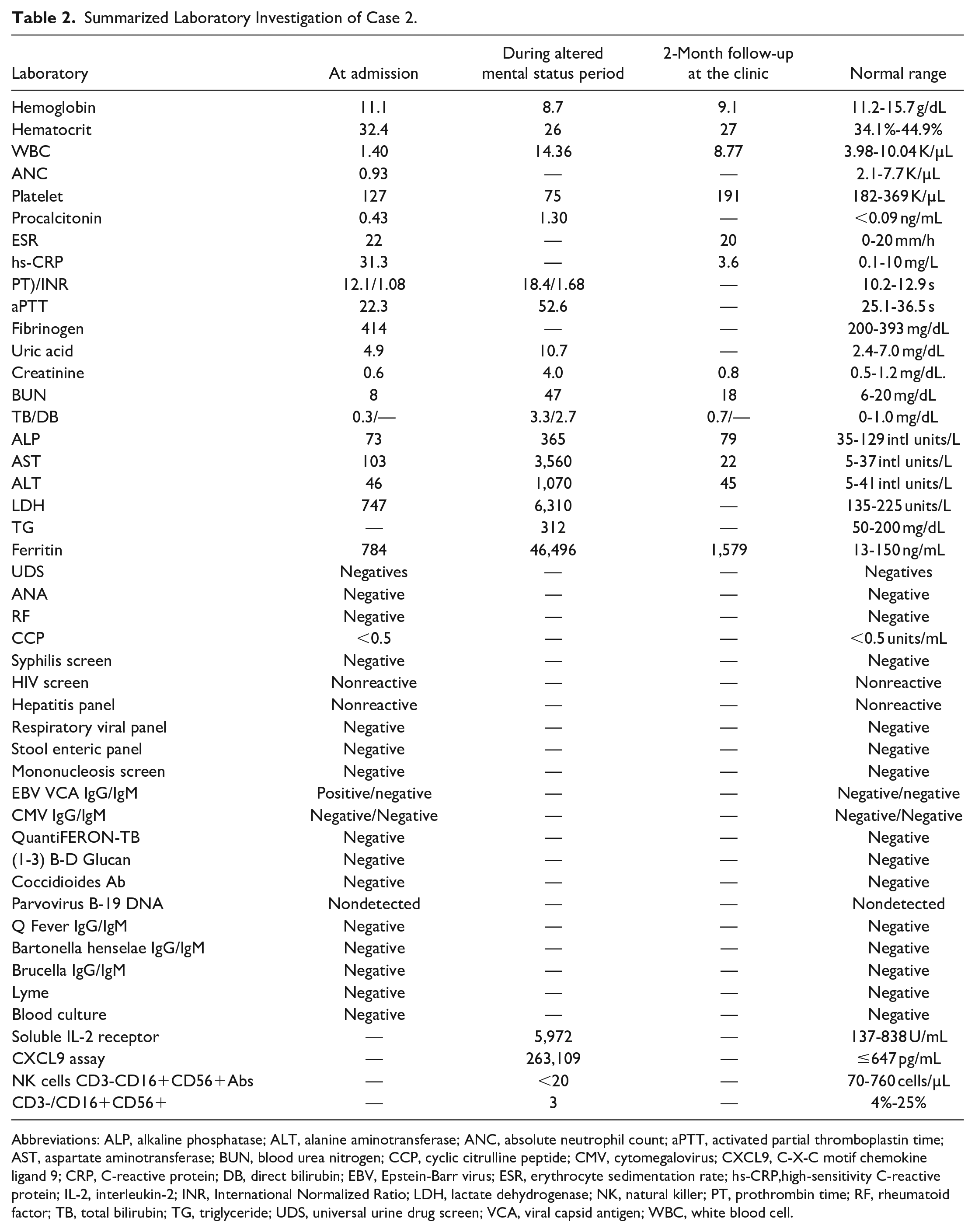
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; ANC, absolute neutrophil count; aPTT, activated partial thromboplastin time; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CCP, cyclic citrulline peptide; CMV, cytomegalovirus; CXCL9, C-X-C motif chemokine ligand 9; CRP, C-reactive protein; DB, direct bilirubin; EBV, Epstein-Barr virus; ESR, erythrocyte sedimentation rate; hs-CRP,high-sensitivity C-reactive protein; IL-2, interleukin-2; INR, International Normalized Ratio; LDH, lactate dehydrogenase; NK, natural killer; PT, prothrombin time; RF, rheumatoid factor; TB, total bilirubin; TG, triglyceride; UDS, universal urine drug screen; VCA, viral capsid antigen; WBC, white blood cell.
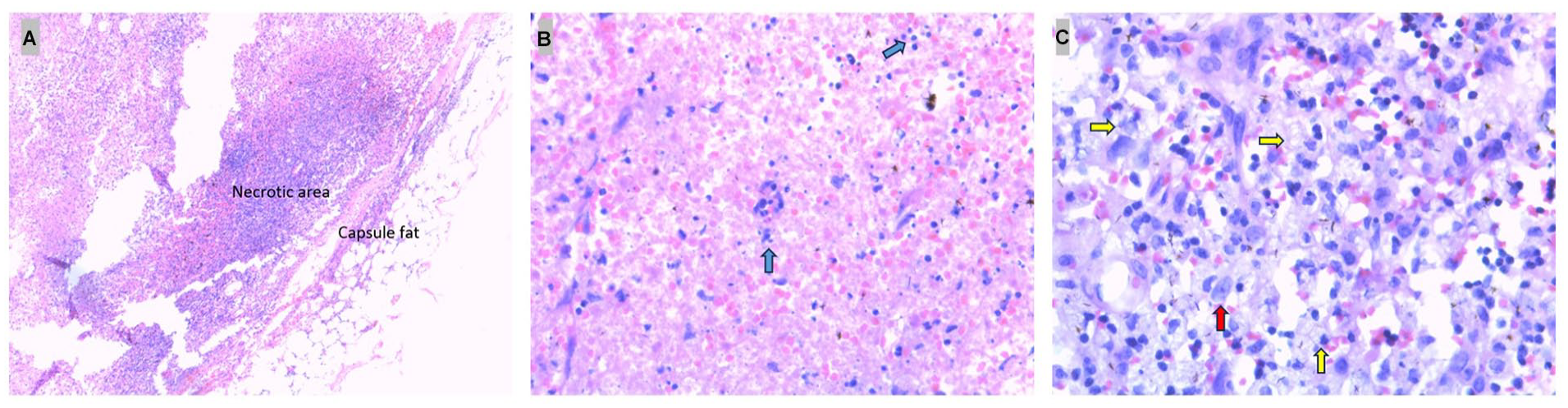
Representative lymph node biopsy (hematoxylin-eosin stain). (A) Lymph node capsule with underlying necrosis, surrounded by histiocytes and dendritic cells (original magnification ×25). (B) Area of necrosis showing karyorrhexis and necrotic nuclei (blue arrows; original magnification ×400). (C) Presence of foamy histiocytes (yellow arrows) and dendritic cells (red arrow; original magnification ×400).
During hospitalization, the patient developed neutropenia (absolute neutrophil count 580 K/µL) with recurrent fever consistent with febrile neutropenia—empirical treatment with broad piperacillin tazobactan, acyclovir, doxycycline, and filgrastim (granulocyte colony-stimulating factor). After 3 doses of filgrastim, the patient developed an altered mental status with an episode of seizure-like symptoms and spontaneous generalized bruising. Repeat laboratory tests showed anemia, thrombocytopenia, coagulopathy, severe transaminitis, hypertriglyceridemia, hypofibrinogenemia, hyperferritinemia, and renal insufficiency (Table 2). A repeat CT of the abdomen and pelvis revealed mild splenomegaly without urinary obstruction. Magnetic resonance imaging of the brain was negative. Cerebral fluid analysis (CSF) demonstrated an elevated CSF protein (89 mg/dL). The repeat bone marrow biopsy was carried out consistent with hemophagocytosis shown in Figure 3. These findings fulfilled 6 out of 8 criteria from the hemophagocytic lymphohistiocytosis (HLH) 2004 diagnostic criteria while waiting for the send-out cytokines profile, with an HScore of 228 indicating a 96% to 98% probability of hemophagocytic syndrome. The diagnosis of MAS secondary to KFD was made. The multidisciplinary team decided to initiate treatment for HLH with dexamethasone (10 mg/m2/day) and etoposide per HLH-94 protocol due to the severity of her laboratory abnormalities and mental status changes. Gradually, the patient showed improvement in symptoms and laboratory values, except for worsening renal insufficiency requiring 1 day of dialysis. She was discharged on day 43 of hospitalization with Anakinra 100 mg subcutaneous injection, mycophenolate mofetil 1500 mg twice a day, and tapering corticosteroid. The soluble interleukin-2 receptor level returned at 5972 (U/mL), meeting the HLH 2004 criteria. She presented for a 2-month follow-up at rheumatology with improved clinical and laboratory values, as shown in Table 2.

Bone marrow biopsy stained with Wright Giemsa. The presence of hemophagocytosis is illustrated, with the black arrow pointing to macrophages containing ingested nucleated red blood cells and mature red blood cells (A and B). Iron stains highlight the engulfed iron particles (C).
Discussion
KFD is a rare condition that can affect individuals across various ethnic groups, primarily presenting in young women. It typically manifests with cervical lymphadenopathy, particularly in the posterior, supraclavicular, or axillary regions, while generalized lymphadenopathy is uncommon. Affected lymph nodes are tender and painful and are associated with fever, weight loss, arthralgia, night sweats, headache, and nausea/vomiting. Hepatomegaly and splenomegaly rarely occur. The onset is typically subacute or acute. Laboratory findings can range from unremarkable to cytopenia, especially anemia and leukopenia, elevated inflammatory markers, and elevated liver function.3,4 The proposed pathological mechanism of KFD is an excessive T cell and histiocytes to different antigens in people with genetic susceptibility.1-3 Patients with KFD are more likely to have certain human leukocyte antigen (HLA) class II alleles, particularly HLA-DPA1 and HLA-DPB1. 3 KFD is believed to be a 2-step disease that requires both a genetic factor and a secondary trigger, such as an infection, to cause an exaggerated T-cell mediated immune and inflammatory response. 1 This syndrome has been associated with autoimmune diseases, particularly SLE, noninfectious inflammatory diseases, and viral infections. 5 The diagnosis of KFD is primarily based on histopathological evaluation. Although excisional biopsy has traditionally been recommended, fine needle aspiration (FNA) from experts generally has an estimated accuracy of 56%. At present, ultrasound-guided core-needle biopsy is preferred, providing a much higher diagnostic accuracy of 95.6% than FNA. 1 Three evolving histologic patterns have been suggested: proliferative, necrotizing, and xanthomatous. The first proliferative pattern shows an enlarged paracortex with many histiocytes and plasmacytoid dendritic cells, mixed with small lymphocytes and karyorrhectic nuclear debris. The necrotizing phase is mainly identified by the presence of necrosis. In the xanthomatous phase, foamy histiocytes are the most common cells in the lesions regardless of the presence or absence of necrosis. 3
In case 1, our patient developed SLE 1 year after the diagnosis of KFD; SLE is the most common autoimmune disease associated with KFD. 6 A systematic review study shows that KFD-SLE cases were mostly female, with an average age of diagnosis at 34 years old. The ethnic distribution was 50.5% Asian and 34% Caucasian. In 18% of patients, SLE was diagnosed before KFD: In 51%, both were diagnosed at the same time, and in 31%, SLE was diagnosed after KFD. No significant clinical differences were found regarding the timing of diagnosis. 7 Many case reports suggest that KFD shares immunopathological and clinical features with SLE. They proposed that KFD may be related to SLE or represent a self-limiting, infection-triggered, SLE-like autoimmune condition.1,8 Typically, KFD requires no special treatment. However, immunosuppressants should be considered when systemic symptoms are severe or accompanied by an autoimmune disease. The interplay between KFD and SLE remains unclear requiring further investigation. 1
Besides the development of autoimmune-associated KFD, a severe complication called MAS has been reported, similar to case 2. MAS is commonly referred to as a secondary form of HLH resulting from autoimmune diseases, characterized by a hyperinflammatory state caused by impaired T-cell degranulation, which prevents the elimination of infected cells. This ongoing stimulation leads to the excessive production of pro-inflammatory mediators, such as interferon-gamma, tumor necrosis factor-alpha, interleukin-1β, interleukin-6, macrophage colony-stimulating factor, and hematopoietic growth factors. 9 Various diagnostic criteria have been implemented, and HLH2004 is widely used. Although the HLH2004 criteria were first developed for pediatric HLH, they are also commonly used for diagnosing HLH in adults. These criteria include 6 clinical and laboratory features: fever, splenomegaly, cytopenias, hyperferritinemia, hypertriglyceridemia/hypofibrinogenemia, and hemophagocytosis. Additionally, there are 3 more specialized tests: genetic testing, natural killer (NK) cell function assessment, and measurement of soluble interleukin-2 receptor (sIL-2r). 10 Among these tests, sIL-2r is the most accessible. 11
Approximately 20 cases of MAS/HLH with KFD have been reported, mostly in teenagers, predominantly male, and 95% are from Asian countries: Korea, Japan, and Taiwan. 12 In Addition, recurrent KFD has the potential to progress to MAS/HLH. Other types of secondary MAS/HLH usually have poor prognosis and high mortality rate, up to 70%, if not diagnosed and treated early.However, KFD with MAS/HLH relatively shows a good prognosis, with complete remission and good response to steroids, IVIG, etoposide, and cyclosporine A. Nonetheless, standardized diagnostic and treatment protocols for this condition have not yet been established.12-15
There is also concern about HLH being aggravated by human granulocyte colony-stimulating factor (G-CSF) analog or filgrastim, as in case 2. The patient’s clinical condition worsened after 3 doses with a suspected temporal relationship. G-CSF is a cytokine that stimulates the production of neutrophils in the bone marrow and improves their function. Based on our knowledge, several cases of G-CSF-induced HLH in patients with hematological conditions have been reported.16-18 Discontinuation of the suspected medication is important for both treatment and prevention in these cases. Previous reports showed that G-CSF-induced HLH can relapse seriously if G-CSF is given again. 16
Conclusion
KFD is an uncommon disease that can occur in various ethnic groups, typically presenting with cervical lymphadenopathy and fever. After excluding cancer, infection, and other autoimmune diseases, early recognition of KFD is crucial to prevent unnecessary further investigation and its complications. Despite its benign course, KFD has been reported alongside other autoimmune diseases, especially SLE and potentially life-threatening conditions. This underscores the need for prompt diagnosis and treatment to reduce morbidity and mortality. To date, treatment guidelines for KFD have not been developed and recommendations are mostly derived from experts’ opinions, case reports, and case series. Further research is needed to improve diagnostic and therapeutic strategies for this rare but significant disease.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethics approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.