
Abstract
Tuberous sclerosis complex (TSC) is a genetic neurocutaneous disorder that presents with multi-organ involvement, including but not limited to hamartomas in the brain, eyes, heart, lung, liver, kidney, and skin. Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory, autoimmune, demyelinating, central nervous system disorder, targeting the optic nerves and spinal cord. We report a 30-year-old woman with TSC who developed tingling in the legs that gradually involved her abdomen. Additional symptoms included severe vomiting that lasted for a week and spasms in her legs. One month later, she was hospitalized due to difficulty ambulating and tingling in her hands. Magnetic resonance imaging (MRI) of her spine showed longitudinally extensive upper cervical and lower thoracic cord signal changes. MRI scan of her brain showed few non-specific T2 signal changes along with cortical and subcortical tubers. Aquaporin (AQP4) IgG antibody was found to be positive in both serum and cerebrospinal fluid. Accordingly, she was diagnosed with NMOSD, treated with a 5-day course of intravenous steroids, followed by 5 sessions of plasma exchange. After her initial improvement, she was started on rituximab as maintenance therapy. Two years later, she is clinically stable, and her follow-up MRI showed marked improvement.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory demyelinating, autoimmune disease consisting of longitudinal transverse extensive myelitis (LETM) and optic neuritis. Most cases are caused by the aquaporin-4 antibody (AQP4), which causes damage to astrocytes leading to inflammation, demyelination, and degeneration. 1 Tuberous sclerosis complex (TSC) is a multi-systematic, autosomal dominant disorder, caused by a mutation of the TSC1 (encoding hamartin) or TSC2 (encoding tuberin) genes. It presents with epilepsy, autism, mental retardation, and non-malignant tumors in any organ. 2
Case Presentation
A 30-year-old woman with TSC, diagnosed by the 2012 International TSC diagnostic criteria, 3 was hospitalized with slight difficulty ambulating and tingling in her hands and lower abdomen. Her diagnosis of TSC was made several years ago, and it was based on facial angiofibromas, several subependymal nodules, and multiple bilateral renal cysts. Her current symptoms started 1 month prior to her hospitalization with severe vomiting for a week and tingling in her legs that spread to her abdomen. One week prior to hospital admission, she started having slight balance impairment and episodic spasms in her arms and legs. No sphincter abnormalities were reported. No visual changes.
Past medical history was significant for TSC and epilepsy which was well-controlled on carbamazepine with last seizure being 5 years ago. She also had a recent uncomplicated fibroid ablation. No recent infection, hospitalization, or metabolic abnormality was reported. No family history of neurological conditions or autoimmune diseases. Social history was negative for alcohol, smoking, and illicit drug use. Patient reported mild urinary frequency and urgency. She denied oral or ocular dryness, rash, arthralgia, weight changes, fever, oral or genital ulcers, photosensitivity, Raynaud’s phenomenon, fatigue, cognitive changes, easy bruising, and hair loss.
Physical examination showed facial angiofibromas; otherwise, it was unremarkable. Neither lymphadenopathy nor salivary gland swelling was noted. Neurological examination showed intact mental function and cranial nerves, with no afferent pupillary defect. Fundus examination revealed intact disks with no retinal abnormalities or optic nerve hamartoma. Strength was full. She was hyperreflexic throughout. No pathological reflexes were elicited. A sensory level to pinprick and light touch at T5 was present. Romberg sign was negative. Coordination was intact. Gait was ataxic.
Magnetic resonance imaging (MRI) scan of the brain showed multiple cortical and subcortical tubers and subependymal nodules including a suspected giant cell astrocytoma at the right foramen of Monro with no hydrocephalus, along with few T2 white matter signal changes in the periventricular white matter (Figure 1). MRI scan of the cervical spine revealed an area of high T2 signal associated with edema involving the medulla and the upper cervical cord, extending to C2-3 level (Figure 2). MRI scan of the thoracic spine demonstrated a T2 hyperintense signal from T7 through T10, measuring approximately 5.5 cm in craniocaudal dimension with edema most pronounced at T8. Enhancing lesions were not detected (Figure 3).

Axial T2 FLAIR showed cortical and subcortical hyperintensities within the brain parenchyma involving the left middle frontal gyrus, right superior frontal gyrus, right parietal cortex, left insula and right occipital lobe, and multiple subependymal nodules (A, B). Axial T1 post-contrast (C) showed enhancing subependymal nodule at the right foramen of Monro and smaller ring enhancing subependymal nodule along the posterior periventricular right white matter.

Sagittal T2 of the cervical spine showed an abnormal T2 hyperintense signal associated with edema involving the medulla and the upper cervical cord, extending to C2-3 level (A). Improvement of these signal changes on follow-up scan 2 years later (B).

Sagittal T2 of the thoracic spine showed an abnormal T2 hyperintense signal from T7 through T10, measuring 5.5 cm in craniocaudal dimension. The edematous quality of this lesion is most pronounced at T8. There is a similar T2 hyperintense area within the right hemicord and dorsal cord at level T12.
Differential diagnosis included multiple sclerosis; myelin oligodendrocyte glycoprotein (MOG) syndrome; idiopathic LETM; secondary causes of LETM of metabolic, infectious, autoimmune, and connective tissue etiology, such as Sjogren syndrome (SS); and systematic erythematous lupus (SEL). Given the patient’s diagnosis of TSC, neoplastic entities, such as astrocytoma, were also included in the differential. Blood work was significant for positive anti-Sjogren syndrome type A (SSA), anti-Sjogren syndrome type B (SSB), and antinuclear antibody (ANA), otherwise was unremarkable. Cerebrospinal fluid (CSF) analysis was normal with a borderline value of two unique oligoclonal bands. Infectious (EBV, CMV, HHV6, VZV, West Nile virus, Enterovirus) and paraneoplastic panels (ANNA-1, 2, 3; AGNA-1; CRMP-5; PCA-1, 2, Tr; amphiphysin) in both serum and CSF were unremarkable. Further workup was significant for positive AQP-4 IgG of 1:100000 in serum and 1:256 in CSF (Table 1). MR spectroscopy showed a choline (cho) peak and decreased N-acetyl aspartate (NAA) with a relatively low cho/NAA of 1.1. No lactate peak was identified, supporting demyelinating etiology versus neoplastic. Despite treatment with steroids, her symptoms progressed to right-sided weakness and significant balance impairment. Subsequently, 5 sessions of plasma exchange were administered successfully.
Serum and Cerebrospinal fluid results
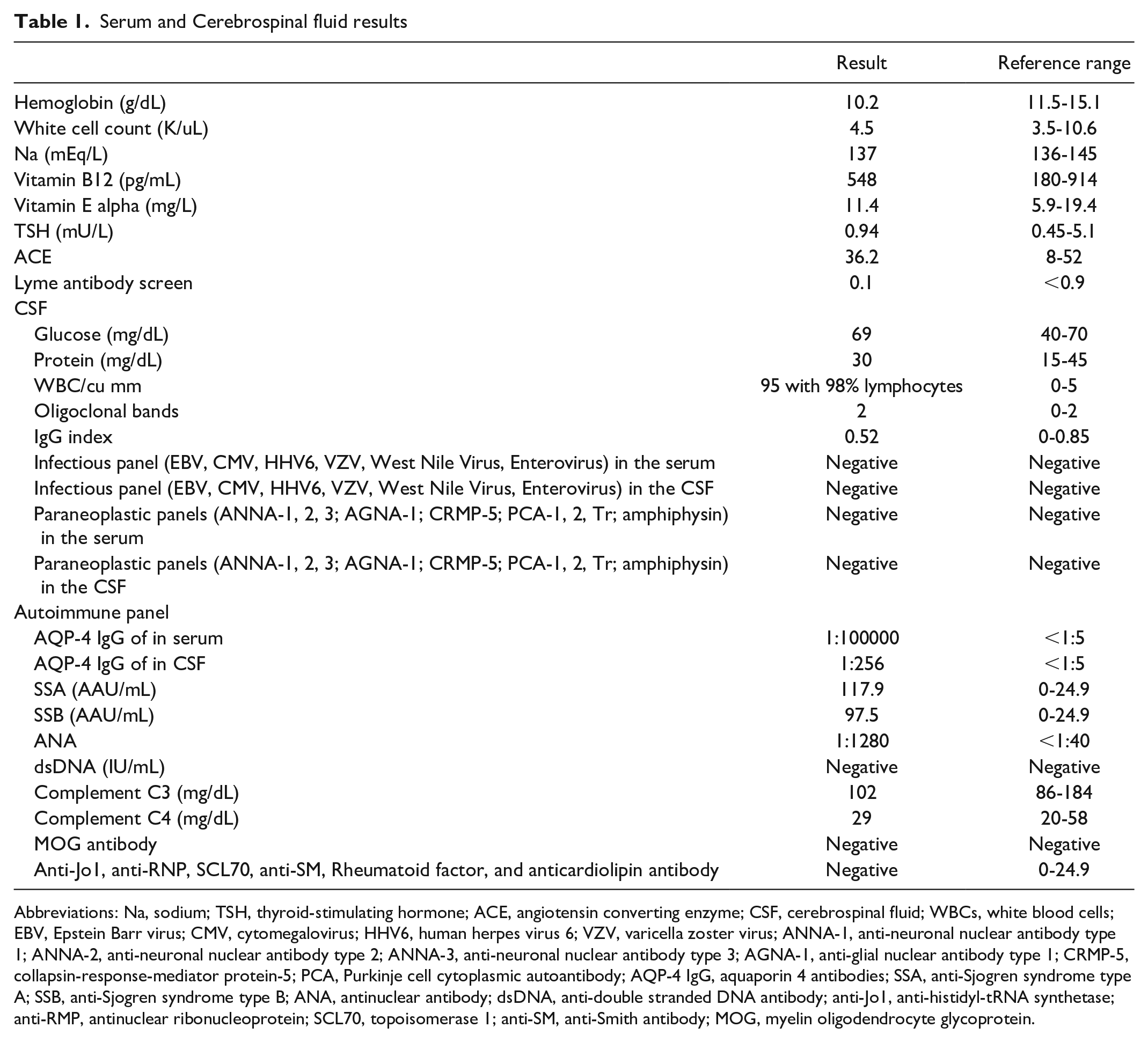
Abbreviations: Na, sodium; TSH, thyroid-stimulating hormone; ACE, angiotensin converting enzyme; CSF, cerebrospinal fluid; WBCs, white blood cells; EBV, Epstein Barr virus; CMV, cytomegalovirus; HHV6, human herpes virus 6; VZV, varicella zoster virus; ANNA-1, anti-neuronal nuclear antibody type 1; ANNA-2, anti-neuronal nuclear antibody type 2; ANNA-3, anti-neuronal nuclear antibody type 3; AGNA-1, anti-glial nuclear antibody type 1; CRMP-5, collapsin-response-mediator protein-5; PCA, Purkinje cell cytoplasmic autoantibody; AQP-4 IgG, aquaporin 4 antibodies; SSA, anti-Sjogren syndrome type A; SSB, anti-Sjogren syndrome type B; ANA, antinuclear antibody; dsDNA, anti-double stranded DNA antibody; anti-Jo1, anti-histidyl-tRNA synthetase; anti-RMP, antinuclear ribonucleoprotein; SCL70, topoisomerase 1; anti-SM, anti-Smith antibody; MOG, myelin oligodendrocyte glycoprotein.
Patient fulfilled the 2015 international panel diagnostic criteria for NMOSD and was started on rituximab. 1 She received 2 doses intravenously of 1 gr each 2 weeks apart, followed by 1 gr every 6 months. She continued her current treatment and showed significant improvement on follow-up visits.
Discussion
Despite the abundance of MRI findings in TSC, such as focal dysplasias, hamartomas, subependymal nodules, and widespread gray and white matter signal changes, spinal cord abnormalities are rarely seen. 4 In our patient, extensive involvement of both cervical and thoracic cord was noted which led to the diagnosis of NMOSD, supported by the presence of AQP4-IgG antibody. Although the association of NMOSD with other autoimmune diseases has been extensively studied, its concurrence with TSC is relatively less elucidated. There are few case reports describing a coexistence of TSC and systemic lupus erythematosus (SLE). Singh and colleagues 5 reported a 26-year-old woman with TSC who developed aggressive SEL with acute and severe renal impairment, not responding to therapeutic interventions. Psarelis and Nikiphorou 6 reported a 26-year-old asymptomatic woman with TSC, who was referred to rheumatology service after detecting a high anti-ds DNA titer. Moreover, Bekkink and colleagues 7 described the rare combination of SLE, TSC, and fulminant natural killer-cell (NK) leukemia in a 39-year-old woman leading to a fatal outcome.
In TSC, mutations of the TSC1 and TSC2 genes result in an abnormally activated mammalian target of rapamycin pathway (mTOR), one of the main pathways regulating T and B cell development, differentiation, metabolism, and function. Subsequently, dysregulation of B and T lymphocytes may lead to abnormal responses and increase the risk for autoimmunity.2,8 Furthermore, Short and colleagues 9 demonstrated increased AQP4-IgG expression by dysfunctional astrocytes in both TSC epileptogenic cortex and astrocyte culture models. As AQP4-IgG is a crucial antibody for the pathogenesis of NMOSD, it is tempting to speculate that the risk for NMOSD may increase in TSC population. 8
Interestingly, our patient had positive SSA and SSB, even though she did not meet the diagnostic criteria for SS. Several investigators reported a high prevalence of SSA and SSB in NMOSD patients.10-13 In a cohort of 48 seropositive AQP4-IgG NMOSD participants, Park and colleagues 10 reported 18 (37.5%) with concomitant positive SSA antibody. Higher AQP4-IgG seropositivity was observed in SSA seropositive compared with the seronegative patients. 10 Estiasari and colleagues 11 reported the presence of AQP4-IgG among 31.8% SS patients, 85.7% of whom had LETM. After studying 106 AQP4-IgG seropositive NMOSD participants, Kim and colleagues 12 reported one third of them positive for ANA and anti-SSA, suggesting a high degree of autoimmunity in this population. Lin and colleagues 13 concluded that SSA/Ro antibody may be associated with NMOSD activity and disability.
This is the first published report of a young patient with TSC and coexisting seropositive NMOSD, two rare diseases that may lead to severe morbidity and adversely affect quality of life. Although the question regarding a common pathogenic mechanism remains unanswered, further studies may shed light on this potential common pathogenesis characterizing both diseases. Reporting such cases is valuable for expanding our understanding of the interplay between genetics and autoimmunity, raising the awareness of coexistence of both diseases and determining whether TSC might confer a higher risk to develop NMOSD.
Footnotes
Acknowledgements
We are thankful to our patient and her family.
Author Contributions
All authors contributed to the design of this case report and were involved in final editing. All authors approve of the final manuscript. M.E. and J.R. share an equal amount of work. E.B. is the corresponding author.
Authors’ Note
Prior presentation of abstract statement: Part of this work was presented as a poster in the Consortium of MS Centers (CMSC) meeting in 2020, which was held virtually, and the relevant abstract was published in the official journal of the meeting, the International Journal of MS Care, 2020, V22, Issue 52, page 84.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series. Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Informed Consent
Verbal informed consent was obtained from the patient for their anonymized information to be published in this article.