
Abstract
Neuromyelitis optica spectrum disorders (NMOSDs) are a set of demyelinating disorders that primarily target the optic nerves and the spinal cord. Previously thought to be a subset of multiple sclerosis (MS), now is recognized as a distinct entity. We present a 59-year-old female patient who was admitted for acute upper and lower extremity weakness. The patient had woken up from sleep with sudden onset of weakness. Patient was initially diagnosed with a right hemispheric stroke; however, magnetic resonance imaging of the cervical spine later performed showed abnormal enhancement from C2-C4, representing transverse myelitis. Cerebrospinal fluid was negative for organisms and inflammatory biomarkers. An anti-aquaporin-4 receptor antibody titer was found to be elevated with titers >80 units/mL. The patient was treated with high-dose steroids and plasmapheresis. The NMOSD is a rare entity and, here, we present a rare presentation of the disease. Since its description in 1870, it was confused with MS for years. The advent of anti-aquaporin-4 antibody has been instrumental in differentiating the disease process from MS. This distinction is important, in terms of agents used for treatment and prognostication. The NMOSD is a set of debilitating disease, which requires prompt recognition and appropriate treatment, to avoid the disabling sequelae. Future prospects of the disease include development of novel biological treatment modalities which focus on restoring the loss of immune tolerance which is key to the pathogenesis of the disease.
Introduction
Neuromyelitis optica spectrum disorders (NMOSDs) are inflammatory disorders of the central nervous system characterized by simultaneous or recurrent immune mediated demyelination and axonal damage to the optic nerves and the spinal cord. The average age of onset is 41 years with a female to male ratio of 6.5:1. 1 The patients typically present with acute attacks of optic neuritis and/or transverse myelitis. Initially, they were thought to be subtypes of multiple sclerosis (MS); however, they are now recognized as separate entities based on pathogenesis, imaging, biomarkers, and response to treatment.
Case Description
A 59-year-old woman with past medical history significant for hypertension, hyperlipidemia, and vertigo was admitted for left upper and lower extremity weakness and left shoulder pain for 2 days. She denied trauma, recent infection, paresthesia, cramps, facial droop, aphasia, urinary, or bowel incontinence, and there were no constitutional symptoms. Her history was negative for malignancy and autoimmune disorders.
On examination, the patient had a heart rate of 83, respiratory rate of 16, blood pressure of 139/96, and an oxygen saturation of 96% on room air. Neurological examination showed normal mentation but unsteady gait. Cranial nerves 2 to 12 were grossly intact. Motor strength was 5/5 in right upper and lower extremities, but 4/5 in left upper and lower extremities. Sensation was intact bilaterally to light touch. Finger-to-nose was dysmetric on the left. Biceps and patellar reflexes were 1/4 on the left and 2/4 on the right. Babinski reflex was absent and Hoffman reflex was present on the left. Given focal neurological findings, magnetic resonance imaging (MRI) of the brain, MRI of the cervical spine (Figures 1 and 2), and lumbar puncture (LP) were performed. Magnetic resonance imaging RI of the brain with and without contrast showed no acute intracranial process.
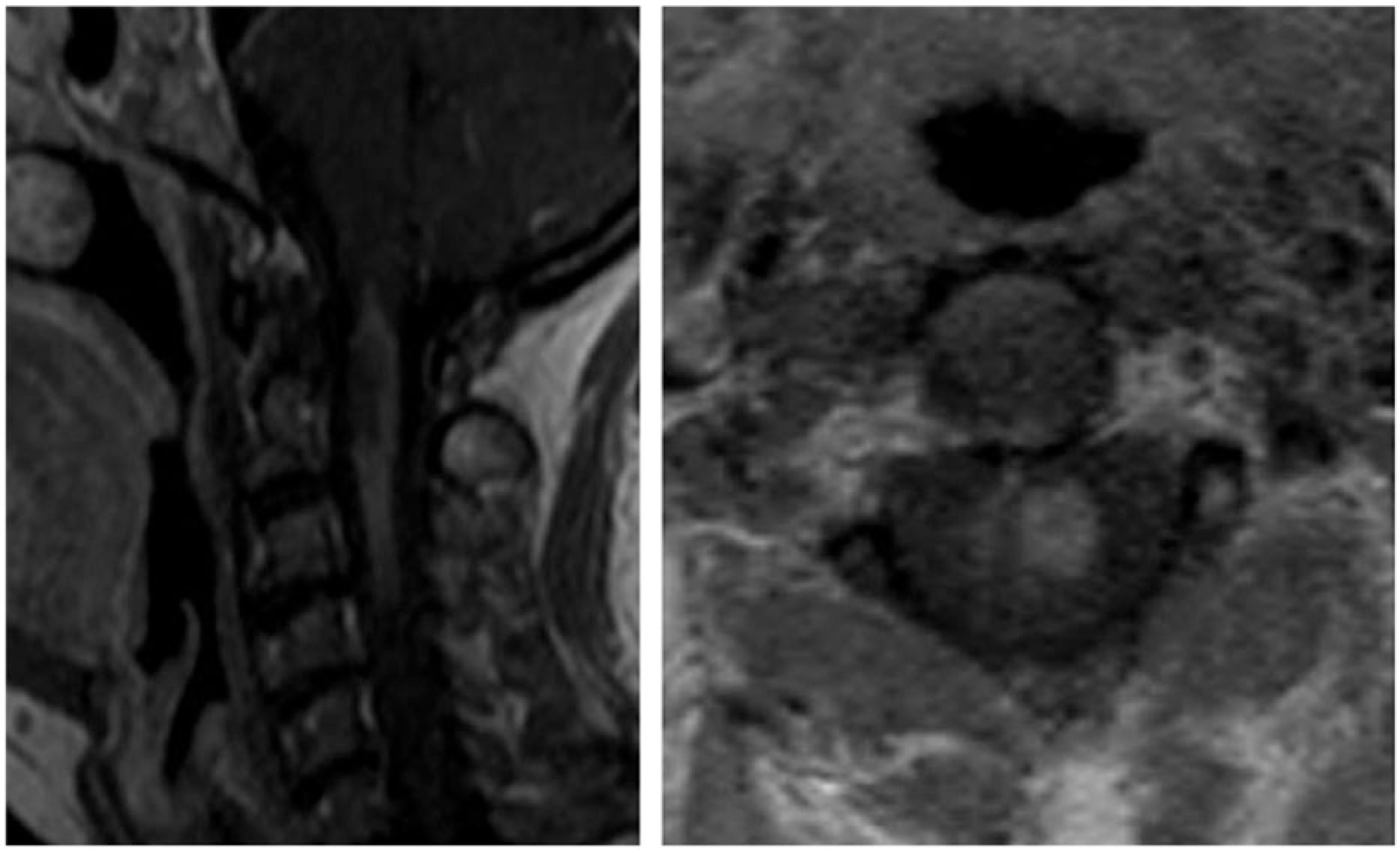
Postcontrast sagittal and axial views of the cervical spine. Read as a focal area of abnormal enhancement within the spinal cord extending from the cranio-cervical junction at level of C3-C4, extending for a length of approximately 5 cm.

Repeat postcontrast sagittal and axial views of the cervical spine, 4 months after previous magnetic resonance imaging. Read as abnormal signal intensity in the left side of the cord without enhancement.
Lumbar puncture showed pleocytosis with lymphocytic predominance and a normal glucose and protein. Cerebrospinal fluid was negative for oligoclonal bands, organisms, neoplastic cells, and inflammatory biomarkers. Serologic testing was negative for cytomegalovirus, human immunodeficiency virus, Lyme, syphilis, and herpes simplex virus. Based on MRI and LP findings, there was suspicion for MS or transverse myelitis causing symptoms, albeit an atypical presentation. Neoplasm or vasculitis was considered unlikely. Therefore, an anti-aquaporin-4 (AQP4) receptor antibody titer was ordered and was found to be elevated with titers >80 units/mL. This is a very disease-specific autoantibody for neuromyelitis optica (NMO) and thus led us to the diagnosis of NMOSD.
The patient was treated with methylprednisolone 1 g intravenous daily for 3 days followed by plasmapheresis for 6 sessions. She was discharged with neurology follow-up outpatient.
Discussion
Neuromyelitis optica, also known as Devic’s disease, is an autoimmune demyelinating disorder of the central nervous system. The disorder was first described in 1870 and was confused with MS for many years. The distinction between NMO and MS is made based on pathogenesis, antibody studies, imaging, and response to treatment. The advent of NMO-IgG/AQP4 was pivotal in differentiating the 2 disease entities. This distinction is important not only for nomenclature but also for therapeutic decisions as some immunotherapeutic agents used for treatment of MS can aggravate the symptoms of NMO. 2
The International Panel for NMO Diagnosis (IPND) revised diagnostic criteria which defined the term NMOSD. The term NMOSD was introduced in 2007 to include patients presenting with atypical form of the disease and were positive for AQP4-IgG serology, those with AQP-IgG seropositivity and concomitant autoimmune disorders, and those with cerebral, diencephalic, and brainstem lesions occurring in patients with typical NMO. In 2015, IPND unified the term NMO and NMOSD and developed the new diagnostic criteria of NMOSD based on the occurrence of core clinical characteristics with or without detection of AQP4 antibody. 3 The core clinical characteristics include the following: (1) optic neuritis, (2) acute myelitis, (3) area postrema syndrome, (4) acute brainstem syndrome, (5) symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions, and (6) symptomatic cerebral syndrome with NMOSD-typical brain lesions. 3 Patients without AQP4-IgG seropositivity have more strict criteria which include 2 or more core clinical characteristics and specific MRI characteristics. A longitudinally extensive transverse myelitis (LETM) extending over 3 or more vertebral segments, as in our patient, is the most specific neuroimaging characteristic of NMOSD and very uncommon in patients with MS. 4 The application of the 2015 criteria was studied by Hamid et al in patients with NMO and non-MS demyelinating disorders from 2006 to 2015. They noted an increase in the diagnosis of NMOSD by 76%. This increase was largely due to the introduction of AQP4 serology testing. 5
In 2006, serology testing for AQP4 was incorporated into the diagnostic criteria for NMO to improve diagnostic accuracy. 4 The AQP4 serum autoantibody, also known as NMO-IgG, has a sensitivity of 91% and specificity of 100% for NMO and is positive in up to 80% of patients. 3 The AQP4 is a water channel in the brain, which is highly localized in astrocytes. These antibodies may be responsible for a variety of immune events which includes immunoglobulin deposition, demyelination, and complement-mediated cytotoxicity. 6 The development of new antigenic targets such as AQP1 and myelin oligodendrocyte glycoprotein in AQP4-seronegative patients may allow for novel treatment options. 7
Acute attacks are treated with intravenous glucocorticoids, followed by plasma exchange for refractory cases, such as our patient. Recurrent attacks are treated with long-term immunosuppression. There are multiple empiric immune-modulating therapeutic options employed. However, none have been proven effective in prospective trials or led to the development of complicated infections. 8 Immunosuppression is continued for at least 5 years in seropositive patients presenting with an initial attack, as they are at high risk for relapse. At this time, there is no consensus as to the total duration of therapy.
The recommended agents include oral azathioprine or mycophenolate mofetil, with or without oral steroids. 9 At present, seronegative and seropositive NMOSDs are managed similarly. The complement fixation pathway, which includes antibody formation and activating the membrane attack complex, plays a key role in NMOSD relapses. Thus, restoring the loss of immune tolerance is fundamental in developing novel NMOSD treatment modalities. 10 New therapies such as anti-IL6 receptor, anticomplement, and anti-AQP4 antibody biologicals are promising future options. 11
The long-term disability and mortality rates of NMOSD are high and there is a stepwise deterioration of function. A retrospective review by Kleiter et al 12 demonstrated that remission rates were higher in isolated optic neuritis as compared with isolated myelitis and for unilateral versus bilateral optic neuritis. 12 Factors indicative of a poor prognosis include the number of relapses in the first 2 years, the severity of first attack, presence of other autoimmune conditions, and older age at onset. 11 Neurogenic respiratory failure is most frequently the cause of death. The use of AQP4 autoantibody as a marker of disease progression is presently being studied but no data are available to suggest its appropriateness in current practice. Weinshenker et al performed a prospective study with patients who initially presented with LETM. Of 29 patients, 11 were found to be seropositive and at 1-year follow-up, 5 of the 9 seropositive patients developed a recurrence. Of the 14 patients who were seronegative at diagnosis, there were no reported recurrences or case reports of optic neuritis. 13
Our patient presented with a rare manifestation of NMOSD, that is, older age of onset without ophthalmologic symptoms. Herein, we highlight the importance of recognizing unusual presentations of NMOSD and the need for early identification and treatment.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
NS was the lead resident physician on the case. AS was the resident physician on the Neurology consult service. SS was the Attending of Internal Medicine and Admitting Physician. IC-N was the Attending Neurology Consultant.