
Abstract
Introduction. According to the 2015 diagnostic criteria for neuromyelitis optica spectrum disorders (NMOSD), in aquaporin-4 immunoglobulin G (AQP4-IgG) seronegative patients, NMOSD can be diagnosed if stringent clinical and magnetic resonance imaging (MRI) criteria are fulfilled; however, in these cases, diagnostic and therapeutic challenges could arise. Case Description. A young man presented a severe, bilateral optic neuritis and paraparesis in an acute phase. MRI evidenced 3 spinal cord T1-contrast-enhanced (T1-Gd+) myelitic lesions, extending <3 vertebral segments. AQP4-IgG and oligoclonal bands were negative. Following a relapse, MRI showed 2 T1-Gd+ spinal cord lesions, extending <3 vertebral segments, and a T1-Gd+ lesion in both optic nerves, near their confluence in the chiasm. After administering rituximab, there were no new relapses, with the consequent improvement of the clinical and MRI lesions. Conclusion. The concurrent display of bilateral optic neuritis and short myelitis constitutes a “borderline” case. Rituximab may represent the most appropriate therapeutic choice possible cases of NMOSD.
Introduction
Neuromyelitis optica spectrum disorders (NMOSDs) are autoimmune inflammatory diseases of the central nervous system (CNS). The prevalence of these diseases ranges from 2.6 to 3.6 cases per 105 in Asians and from 0.7 to 4 cases per 105 in Caucasians, and the median age of onset is 39 years. 1 The most typical clinical features of NMOSD are optic neuritis (ON) and acute transverse myelitis. Less common clinical features could be area postrema syndromes, encephalopathy, and so on. 1 A second relapse occurs within 1 year in 60% of patients and within 3 years in 90% of the cases, leading to severe disability. 1 Serum aquaporin-4 immunoglobulin G (AQP4-IgG) is a reliable and potentially pathogenic biomarker of NMOSD (73% sensitivity, by cell-based assays, and 91% specificity). However, 10% to 25% of NMOSD patients are seronegative for AQP4-IgG (AQP4-IgGneg). 1 The 2015 diagnostic criteria 2 enable the detection of NMOSD in seropositive AQP4-IgG (AQP4-IgGpos) patients through the analysis of almost any CNS region. For AQP4-IgGneg patients, detailed clinical, neuroimaging, and laboratory analyses are required to increase diagnostic accuracy, because these patients could be affected by multiple sclerosis (MS)/overlapping syndromes (eg, acute disseminated encephalomyelitis, neuro-systemic lupus erythematosus, Sjögren syndrome, neuro-Behçet disease, neurosarcoidosis, paraneoplastic and autoimmune encephalitis). An accurate diagnosis of such disorders has important treatment implications, because some MS immunotherapies could worsen NMOSD. 2 Immunosuppressive agents are the best treatment strategy when there is suspicion of NMOSD, and rituximab, among others, is increasingly recognized as an established therapy for NMOSD, with long-term efficacy and an acceptable safety profile.3,4 The purpose of this case report is to show how difficult it can be to diagnose NMOSD based on stringent diagnostic criteria, and how it can be necessary to make a diagnosis after a careful differential diagnosis based on the typical clinical-radiological presentation of the disorder, in order to administer the most effective treatment possible with the lower risk of side effects.
Case Presentation
We report a case of a 35-year-old man of Asian Indian ethnicity, born in Bangladesh and resident in Italy since 2001. No relevant diseases were detected in his past medical history. He had a family history of type 1 diabetes mellitus (father). He had worked as a painter for over 10 years, with possible exposure to toxic substances. However, he reported to have used protective air masks. On day 1, he reported frontal and binocular pain, and he had a drastic decline in visual acuity. He was hospitalized in the Ophthalmology Unit, where brain computed tomography scan, retinal fluorescein angiography, and standard bloodwork (kidney and liver function, fasting glucose, blood count, inflammatory markers, serum proteins, iron, vitamin B12, folate, and urinalysis) were carried out with normal results. Blood lead and trichloroethanol, as well as urinary methanol, trichloroacetic acid, and lead were undetectable. Signs of an existing systemic infectious disease were absent (Table 1). The analysis of visual-evoked potentials showed P100 waves of bilateral higher latency and amplitude at lower limit. A diagnosis of bilateral ON was performed, and a high-dose steroid treatment (intravenous methylprednisolone 1 g per day for 5 consecutive days) was started on day 3, with no improvement of visual acuity. Two days after the administration of high-dose steroids (day 5), the patient developed a rapidly worsening paraparesis, associated with bladder dysfunction. A brain and total (cervical, thoracic, lumbar, sacral segments) spinal cord (SC) magnetic resonance imaging (MRI) was performed. It showed the following: a T2-hyperintense, T1-isointense, T1-non-constrast-enhanced (T1-Gd−), and nonedematous lesion (8 mm) in the left parietal subcortical white matter (WM); other small, nonspecific, T2-hyperintense, and T1-Gd− lesions in the subcortical WM; 3 SC T2-hyperintense, T1-isointense, and T1-constrast-enhanced (T1-Gd+) lesions, with an oval morphology, an axial central position, and extension <3 vertebral segments (VSs; located in the thoracic SC, posteriorly to the T1-T2, T5, and T6 VSs; Figure 1a-g). On day 20, the patient was transferred to the Neurology Unit. The neurological examination showed that the gait was paraparetic (worse at the right lower limb) and was possible for only a few steps with monolateral assistance; the strength against maximum effort was reduced in all movements made with the lower limbs, more on the right than on the left side; a mild spasticity was present in the right lower limb; osteo-tendon reflexes were evocable with greater excitability in the right lower limb (with an evocable clonus at right ankle); a Babinski’s sign was present bilaterally; visual acuity was reduced to hand motion in both eyes; there were no deficits of orientation, cognition, motor functions, and osteo-tendon reflexes in the upper limbs, as well as in sensory, cerebellar, and other cranial nerve functions. Cerebrospinal fluid analysis showed a moderate increase of protein level (71 mg/dL), a normal cell count (4 cell/mm3), and the absence of oligoclonal bands (OBs). Signs of existing infectious disease of the CNS were absent (Table 1). Other autoimmune diseases (eg, acute disseminated encephalomyelitis, systemic lupus erythematosus, Sjögren syndrome, neuro-Behçet disease, sarcoidosis, primary angiitis of the CNS, paraneoplastic and autoimmune encephalitis) were excluded because of the absence of typical clinical, humoral, and radiological (MRI, chest X-ray) findings (Table 2). Serum neoplastic markers were in their normal ranges. Both serum myelin oligodendrocyte glycoprotein-IgG and AQP4-IgG (tested by cell-based assay) were negative; this result was confirmed also in a second analysis repeated 1 month later in a different laboratory. A second high-dose steroid treatment was started on day 20, with partial improvement of the paraparesis, but no effect on the visual deficit. On day 31, the patient’s visual acuity, bladder function, and paraparesis worsened, the patient could no longer ambulate, and there were also signs of bilateral hypesthesia, with a T4 sensory level, worse on the left side. A treatment with 1 cycle of therapeutic plasma exchange was started, with partial improvement of only visual and bladder symptoms. A new MRI of the brain, orbits, and total SC showed the following: a significant enlargement (23 mm) with ring contrast enhancement and T1-hypointensity of the previous left parietal subcortical WM lesion (Figure 2a and b); ring contrast enhancement of the thoracic SC lesion located posteriorly to the T1-T2 VSs, and confluence of the thoracic SC lesions located posteriorly to the T5 and T6 VSs in a single thoracic SC lesion located posteriorly to the T4-T6 VSs (<3 VSs), with increase in extension, T1-isointensity, and T1 ring contrast enhancement (Figure 2c and d); a tumefactive T2-hyperintense and T1-Gd+ lesion in both posterior optic nerves, near their confluence in the chiasm, mostly on the right side (Figure 2e). A third steroid cycle was started on day 39, followed by a significant improvement of the paraparesis, and a partial but promising effect on sensitive, visual, and bladder functions. In the days that followed, the patient started neurorehabilitation, and his gait progressively improved with bilateral assistance. Subsequently, a prophylactic treatment with rituximab was chosen and started with 1 g infusion on day 68 (month 2), followed by a second 1 g infusion 2 weeks later (day 82, month 3), with subsequent suppression of circulating CD19+ B-cell to 0%. Re-treatment was then planned after 6 to 9 months. Nine months later (month 13), since CD19+ B-cells levels were 2%, a third 1 g infusion was performed. Ten months later, CD19+ B-cells levels were 3% to 4% and a fourth 1 g infusion was performed (month 23). No side effects occurred in any infusion. No more relapses have occurred for almost 3 years, and neurological examination showed a slow progressive improvement, with stability from month 5 until now: the gait is slightly paraparetic and possible without aid or rest >500 m; muscular strength, tone, and osteo-tendon reflexes are all normal; hypesthesia and dysesthesia with T4 sensory level is bilaterally improved; and the visual acuity is still reduced in both eyes but has improved since the onset of the disease. The clinical stabilization was confirmed by repeated MRI scans of the brain and total SC. During the last MRI of the SC, performed in month 13, we observed no new lesions, no contrast enhancements or T1-hypointensity, dimensional reduction of the left parietal subcortical WM lesion, and thickness reduction of the thoracic SC lesion located posteriorly to the T4-T6 VSs. Moreover, no cortical lesions were detected on a double inversion recovery sequence performed in the last MRI scan.
Infectious Diseases Diagnostic Assay.

Abbreviations: S, serum; VDRL, venereal disease research laboratory test; L, cerebrospinal fluid; TPHA, Treponema pallidum Hemagglutination Assay; anti, antibodies; IgM/G, immunoglobulins M/G; HHV, human herpes virus; HSV, herpes simplex virus; VZV, varicella-zoster virus; CMV, cytomegalovirus; JC, John Cunningham virus; HIV, human immunodeficiency virus; HTLV, human T-lymphotropic virus; PCR, polymerase chain reaction; UA, allergenic unit; U, unit; EBV, Epstein-Barr virus; VCA, virus capsid antigen; EBNA, Epstein-Barr nuclear antigen.

Brain and total spinal cord (SC) magnetic resonance imaging (MRI) performed at the onset of the disease, showing the following: a T2-hyperintense lesion (8 mm) in the left parietal subcortical white matter (WM), and other small, nonspecific, T2-hyperintense lesions in the subcortical WM, all of them without T1 contrast enhancement (a-c); 3 thoracic SC T2-hyperintense lesions, with extension <3 vertebral segments (VSs; respectively, located posteriorly to the T1-T2, T5, and T6 VSs) and T1-contrast-enhancement (d-g).
Autoimmune Diseases Diagnostic Assay.

Abbreviations: S, serum; ANA, antinuclear antibodies; anti, antibodies; ENA, extractable nuclear antigens; U1RNP, U1 ribonucleoprotein; Sm, Smith; SSA, Sjögren’s syndrome–related antigen A; SSB, Sjögren’s syndrome–related antigen B; CENP-B, centromere protein B; SCL70, scleroderma antigen; Jo1, anti-histidyl–tRNA synthetase; nDNA, native DNA; ANCA, antineutrophil cytoplasmic antibodies; AMA, antimitochondrial antibodies; ASMA, anti-smooth muscle antibodies; APCA, anti-gastric parietal cell antibodies; ACA, anticardiolipin antibodies; IgM/G, immunoglobulins M/G; TPO, thyroid peroxidase; TG, thyroglobulin; ACE, angiotensin converting enzyme.

Brain and total spinal cord (SC) magnetic resonance imaging (MRI) performed at the relapse of the disease, showing the following: significant enlargement (23 mm) of the previous left parietal subcortical white matter (WM) lesion (a), with T1 ring contrast enhancement (b) (T1 scans not shown); T1 ring contrast enhancement of the thoracic SC lesion located posteriorly to the T1-T2 vertebral segments (VSs), confluence of the thoracic SC lesions located posteriorly to the T5 and T6 VSs in a single lesion located posteriorly to the T4-T6 VSs, with increase in extension (35 mm, <3 VSs) (c), and T1 ring contrast enhancement (d); a T1 contrast-enhanced lesion in both optic nerves, near their confluence in the chiasm, mostly on the right side (e).
Discussion and Conclusion
After the first clinical event, represented by an acute bilateral ON and a myelitis, making a diagnosis of NMO was not possible according to the 2006 Wingerchuk diagnostic criteria, 5 because of the lack of serum AQP4-IgG and longitudinally extensive transverse myelitis (LETM; Table 3). We could not even make a definite diagnosis of NMOSD according to the 2015 criteria for NMOSD AQP4-IgGneg patients, 2 because the required association between acute myelitis and LETM was not fulfilled, even though, on MRI, both optic nerves presented a T2-hyperintense and T1-Gd+ lesion in their posterior part, involving the optic chiasm (Table 4). Even if possible according to the 2010 McDonald criteria, 6 a diagnosis of MS was unlikely, because of the absence of most MS-typical clinical and radiological features apart from short myelitis (eg, monolateral and mild-to-moderate ON, with spontaneous or steroid-induced recovery of visual acuity; asymptomatic, extending <1 VS, peripheral WM lesions on SC MRI; cortical, periventricular, or juxtacortical WM lesions on brain MRI; type 2-OBs). MRI lesions pattern could help in the differential diagnosis of CNS demyelinating diseases (especially MS). LETM lesions are the most specific neuroimaging marker of NMOSD and are very uncommon in adult MS patients. 5 These lesions are usually symptomatic, with an extension ⩾3 VSs, T1-Gd+, and localized in the central SC gray matter (as NMOSD are currently considered astrocytopathies rather than disorders of myelin) and in the upper thoracic SC segments 7 ; in contrast, MS SC lesions are usually asymptomatic, with an extension ⩽1 VS, T1-Gd−, and localized in the peripheral WM and in the cervical SC segments.2,7 The timing of MRI scan may be very important for the demonstration of LETM: in fact, lesions extending <3 VSs could be detected if MRI is performed too early or too late in the evolution of acute myelitis, or after immunosuppressive treatment, because a LETM lesion may fragment into multiple shorter lesions.2,8 Consequently, the MRI scan must be carried out as soon as possible after the onset of the first symptoms and before initiating an immunosuppressive treatment. It is likely that this factor could have limited the detection of LETM in the MRI of our patient. Unilateral or bilateral increased T2-signal or T1-gadolinium enhancement within the optic nerve or optic chiasm, relatively long lesions (ie, extending more than half the distance from orbit to chiasm), together with the lesions involving the posterior part of the optic nerves or the chiasm are associated with NMOSD. 2 Cortical lesions, detectable by double inversion recovery sequences, are atypical for NMOSD and typical for MS. 2 Nonspecific brain small lesions (<3 mm) and large confluent WM lesions, with tendency to shrink and even disappear, are common findings in NMOSD (35% to 84%). 2 Patients with NMOSD are less likely to develop clinically silent MRI lesions than patients with MS. 7 Moreover, most studies show that nonlesional tissue damage (measured by diffusion tensor MRI), which is rather common in MS, does not occur in NMOSD. This supports the hypothesis that, unlike MS, NMOSD may be a lesion-dependent disease that produces relapses without a progressive clinical phase accompanied by a generalized neurodegeneration. 9 Combinations of NMOSD and MS features (ie, severe ON and short myelitis) are not uncommon in patients suspected for NMOSD. Therefore, it has been proposed that different degrees of diagnostic certainty could be particularly useful in such phenotypes. 10 In this perspective, our patient was more likely to have NMOSD than MS, mainly because of the following features: (1) bilateral and severe ON (almost blindness), with little recovery of visual acuity—which occurs neither spontaneously nor after steroid treatment—and association with a T2-hyperintense and T1-Gd+ lesion in both posterior optic nerves, involving the optic chiasm; (2) myelitis associated to a symptomatic lesion on MRI, which was T1-Gd+, localized in the central SC gray matter and in the upper thoracic SC segments; (3) absence of MS-typical brain cortical, periventricular (“Dawson’s fingers”) or juxtacortical WM lesions, and of SC asymptomatic, extending <1 VS, T1-Gd−, peripheral WM lesions on MRI; (4) absence of MS-typical OBs; (5) absence of MS-typical clinical and radiological dissemination in space and time of the disease. Patients suspected for NMOSD who are AQP4-IgGneg could also represent a diagnostic challenge despite the use of the best available assays. AQP4-IgG levels tend to increase during relapses and to decrease when immunosuppressive drugs are administered. 2 These factors could have limited the detection of AQP4-IgG in the serum of our patient. Dosing AQP4-IgG as closely as possible to the clinical manifestations of a suspected NMOSD, and before initiating an immunosuppressive treatment, could represent a more correct and reliable diagnostic workup. In AQP4-IgGneg patients, the presence of myelin oligodendrocyte glycoprotein-IgG and aquaporin-1 (AQP1)-IgG, which could cause a similar clinical picture, should be ruled out. 1 In our case, the former has been excluded, while the latter has not been tested. AQP4-IgGneg patients tend to have lower relapse rate and lower disability according to many studies. 11 Further research needs to be carried out on the possible differences in neuroimaging features between AQP4-IgGpos and AQP4-IgGneg NMOSD. An induction therapy, made by immunosuppressive agents, is currently considered the best approach to treat inflammatory autoimmune diseases with a high risk of relapses-derived disability, such as NMOSD and MS.10,12 The main treatment objectives for NMOSD are the improvement of relapse-associated symptoms and the prevention of relapses. Therefore, rituximab is increasingly recommended as a first-line treatment for these diseases, 13 even if class I studies are still lacking. 3 Early immunosuppression is fundamental for AQP4-IgGpos and AQP4-IgGneg patients, because AQP4-IgGneg patients risk a conversion to the typical AQP4-IgGpos NMOSD phenotype and, thus, of relapses. To date, a standardized rituximab treatment protocol in NMOSD does not exist, but there is general agreement that an induction therapy with infusion of 1 g of the drug for 2 weeks can provide a deep B-cell depletion and therefore a faster and long-lasting stabilization of the disease. 14 For the maintenance therapy, a fixed reinfusion of 1 g of the drug every 6 to 9 months is recommended in order to obtain 2 to 4 years of disease stability in 81% to 84% of patients,3,13,15 considering that B-cell repletion, and a major relapse risk, takes place after at least 6 months. Monitoring CD19/20+ B-cell count could be a practical way to mark the immunosuppressed state of the patient and the therapeutic efficacy of rituximab. In conclusion, this case report and the related literature suggest that, even in absence of AQP4-IgG and LETM, the concomitant onset of severe bilateral ON and short myelitis (lesions extending <3 VSs), in absence of typical clinical and radiological MS features, should raise a strong suspicion of NMOSD, and that rituximab could be the most effective treatment for these diseases. 4
Proposed Diagnostic Criteria for NMO (2006).

Abbreviations: NMO, neuromyelitis optica; MRI, magnetic resonance imaging; IgG, immunoglobulin G.
NMOSD Diagnostic Criteria for Adult Patients (2015).
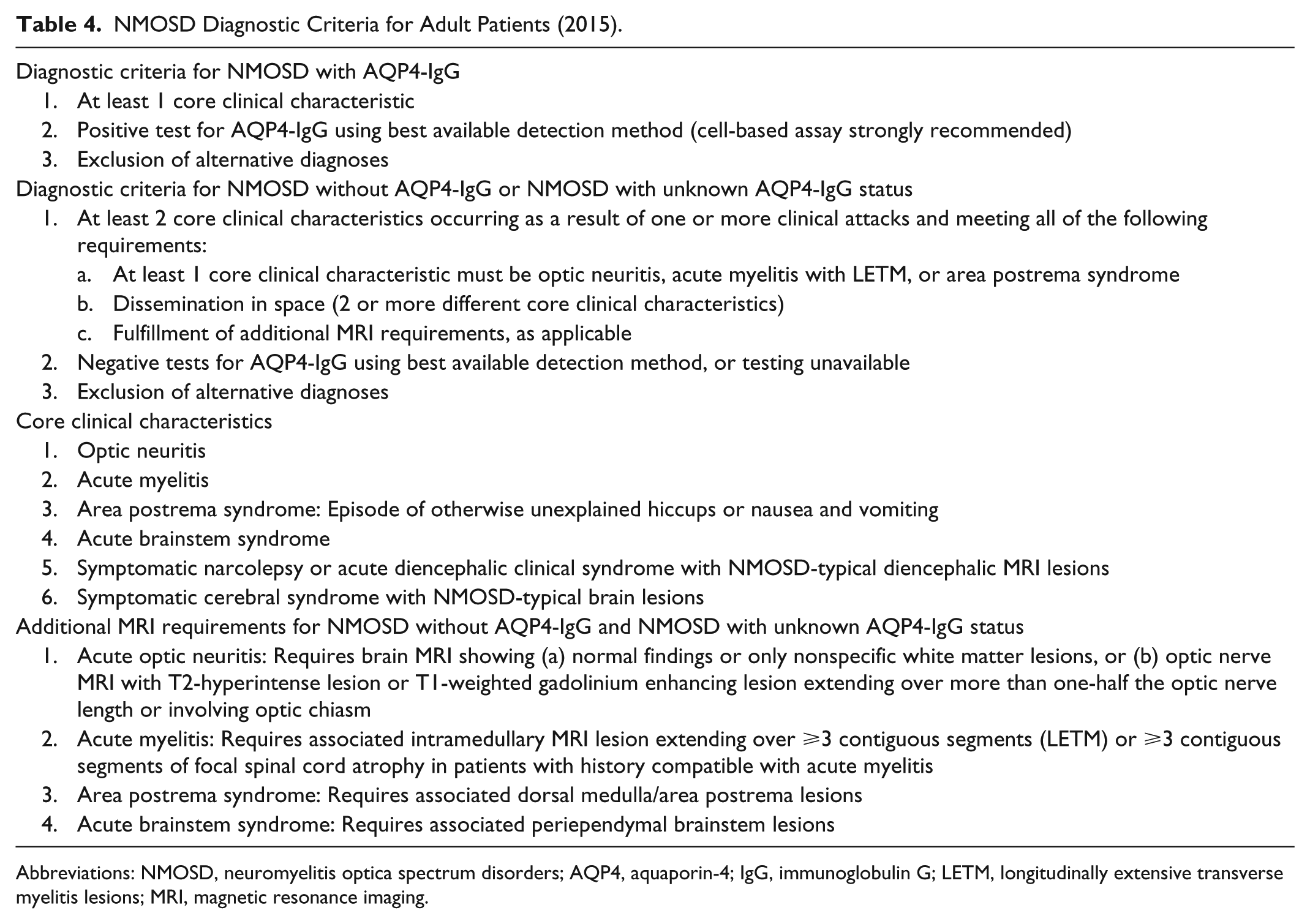
Abbreviations: NMOSD, neuromyelitis optica spectrum disorders; AQP4, aquaporin-4; IgG, immunoglobulin G; LETM, longitudinally extensive transverse myelitis lesions; MRI, magnetic resonance imaging.
Footnotes
Acknowledgements
The authors thank all neurologists and nurses of the Neurology Unit for their support and clinical suggestions, and all physiatrists and physical therapist of the Rehabilitation Unit who took charge of the neurorehabilitation of the patient.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the patient for their anonymized information to be published in this article.