
Abstract
The study aimed to develop and validate a precise, reliable, and sensitive LC–MS/MS method for the simultaneous estimation of Sofosbuvir (SOF), Voxilaprevir (VOX), and Velpatasvir (VEL) in rat plasma for pharmacokinetic applications. Plasma samples were protein-precipitated and analyzed using an Agilent Eclipse XDB C18 column with an acetonitrile–ammonium formate (20:80, pH 3.0) mobile phase, and detection was carried out on a SCIEX QTRAP 5500 in MRM mode. Optimized mass transitions were m/z 530.4537 → 294.1257 for SOF, 869.9463 → 259.6483 for VOX, 884.0635 → 761.3207 for VEL, and 883.0154 → 587.4326 for the internal standard, Elbasvir. The method demonstrated excellent linearity (R2 > 0.999), acceptable precision (%CV ≤ 15%), and accuracy (85%–115%). Recovery of all analytes ranged from 95.39% to 98.74%, with negligible matrix effects. The LLOQ values were 20 ng/mL for SOF, 5 ng/mL for VOX, and 5 ng/mL for VEL. The developed method demonstrated acceptable accuracy, precision, and sensitivity for preclinical pharmacokinetic applications. The method was successfully applied to a rat pharmacokinetic study, indicating its suitability for exploratory bioanalytical investigations.
Introduction
Hepatitis C virus (HCV) infects over 170 million people worldwide and is a major cause of chronic liver disease. 1 The use of antivirals such as Sofosbuvir (SOF), Velpatasvir (VEL), and Voxilaprevir (VOX) has revolutionized HCV therapy, offering pan-genotypic, highly effective, and well-tolerated regimens. 2 The combination of SOF/VEL/VOX represents one of the most potent therapeutic options for direct-acting antivirals (DAAs)-experienced patients, emphasizing the importance of accurate pharmacokinetic and bioanalytical quantification of these drugs in biological matrices.1,2
Extensive research has focused on the analytical and bioanalytical evaluation of these DAAs to ensure their quality, stability, and therapeutic efficacy.3–5 Recent reviews have summarized the evolution of chromatographic and spectroscopic strategies, highlighting a growing trend toward eco-friendly and green analytical methodologies.3,4 Early investigations primarily utilized RP-HPLC and UPLC methods to quantify SOF and VEL in dosage forms.6–13 However, limitations in sensitivity and selectivity prompted the transition toward LC–MS/MS-based bioanalytical platforms, which offered superior accuracy and throughput for plasma quantification.14–16
Among the notable developments, Gandhi et al. 15 developed a sensitive method (LLOQ ≈ 4 ng/mL), which represented a significant advancement in DAA quantification. Subsequent mass spectroscopic methods for binary combinations such as SOF + VEL,14,16,17 and triple combinations (SOF + VEL + VOX) in dosage forms,18–21 demonstrate the growing analytical interest in this antiviral regimen. Stability-indicating RP-HPLC and PDA-based techniques have been optimized for formulation matrices,20–23 but comprehensive protocols for all three drugs in plasma remain limited.
Advanced LC–MS/MS platforms have further improved selectivity and detection limits for SOF and its metabolites.24–26 Moreover, chromatographic advances have supported pharmacokinetic applications across multiple biological matrices (plasma, CSF, seminal fluid),26,27 highlighting their relevance in both antiviral and repurposed therapeutic studies.
Recent analytical trends emphasize method greenness, miniaturization, and automation to improve sensitivity and reduce solvent use.3,4,28–30 Emerging analytical designs using chemometric optimization and response surface methodology (e.g., Box–Behnken designs) have enhanced accuracy and robustness.28,29 Additionally, modern fluorescence- and TLC-based detection approaches have been introduced for the selective estimation of VEL and SOF in plasma, contributing to simplified, cost-effective bioanalysis.31,32
Parallel advances in formulation and stability-indicating methods continue to strengthen pharmaceutical quality assessment, with recent work focusing on HPLC, UFLC, and spectrophotometric quantification for combined antiviral formulations.9–13,18–23,30,33–37 These methods are instrumental for ensuring dosage-form integrity, identifying degradation pathways, and supporting regulatory submissions.
The present study aims to develop and validate an LC–MS/MS method for simultaneous quantification of SOF, VOX, and VEL in rat plasma to support preclinical pharmacokinetic investigations. This optimized approach is intended to support pharmacokinetic profiling, bioequivalence evaluation, and therapeutic drug monitoring in preclinical investigations, while setting a benchmark for future DAA bioanalytical studies.26,27,30–32,34–37 Despite the availability of several analytical methods for the determination of DAAs, most reported approaches focus on single drugs or binary combinations, primarily in pharmaceutical formulations. Reports on simultaneous quantification of SOF, VOX, and VEL in biological matrices remain limited, particularly for preclinical pharmacokinetic applications.
Methodology
Reagents and Chemicals
SOF, VOX, VEL, and the internal standard Elbasvir (IS), each with purity ≥98%, were kindly provided by Biocon Biologics Limited, Bengaluru, India. LC–MS grade ACN and MeOH were obtained from Merck Chemical Division, Mumbai, India, while all other solvents and reagents were of LC–MS or analytical reagent grade.
Chromatographic Conditions
The Waters Alliance HPLC system was coupled to a QTRAP 5500 MS and used an Agilent Eclipse XDB-C18 column (250 × 4.6 mm, 5 µm) with a 20:80 acetonitrile–ammonium formate (adjusted the pH to 3.0 with formic acid) mobile phase. Chromatographic separation was performed at room temperature with a flow rate of 1.0 mL/min, 10 µL injection volume, and a 7-minute run time. The ammonium formate buffer was used as the diluent. Under these optimized conditions, SOF, VOX and VEL were well resolved with sharp, symmetrical peaks, while Elbasvir (IS) served as a stable IS, confirming system suitability and reproducibility (Figure 1).
Representative MRM Chromatogram of Sofosbuvir (SOF), Voxilaprevir (VOX), Velpatasvir (VEL), and Internal Standard Elbasvir Obtained Under Optimized LC–MS/MS Conditions.

Mass Spectrometric Conditions
Quantification was performed on ESI⁺ mode using MRM for each analyte and the IS. The optimized MS parameters are presented in Figure 2. All data acquisition, peak integration, and quantitative analysis were performed using Analyst Software (SCIEX). The optimized mass conditions yielded stable ionization, adequate sensitivity, and minimal background interference for all three analytes. The optimized multiple reaction monitoring (MRM) transitions were as follows: SOF m/z 530.4537 → 294.1257, VOX m/z 869.9463 → 259.6483, VEL m/z 884.0635 → 761.3207, and the IS Elbasvir m/z 883.0154 → 587.4326. The collision energy was maintained at 14 V for all analytes, with a declustering potential of 40 V, entrance potential of 10 V, and exit potential of 7 V. These optimized parameters ensured stable ionization, reproducible fragmentation, and sensitive detection for all analytes.
Optimized MRM Transitions of SOF, VOX, VEL, and Internal Standard Elbasvir (IS).

Preparation of Solutions
Stock solutions of SOF, VOX, and VEL were prepared in ammonium formate buffer (pH 3.0), followed by serial dilutions to obtain working and combined standards. Working standards refer to diluted stock solutions used for calibration preparation, while the combined standard represents a mixture containing all analytes at their respective concentrations. Working standard solutions were prepared at concentrations of 16 µg/mL for SOF and 4 µg/mL each for VOX and VEL. A combined standard solution containing all three analytes at the same respective concentrations was prepared by mixing appropriate aliquots of the individual working standards. The IS (Elbasvir) was similarly prepared at 400 ng/mL. Calibration standards were prepared in rat plasma (SOF: 20–800 ng/mL; VOX/VEL: 5–200 ng/mL) and stored at 2°C–8°C until analysis. The IS (Elbasvir) working solution was prepared at 400 ng/mL and used for spiking into all calibration standards, quality control (QC) samples, and study samples prior to extraction.
Sample Preparation and Extraction Procedure
Plasma sample preparation was carried out using a protein precipitation technique. To ensure accurate quantification and to correct for extraction variability and instrumental fluctuations, the IS (Elbasvir) was added to all samples prior to extraction.
Briefly, 200 µL of rat plasma was transferred into a centrifuge tube, followed by the addition of 50 µL of IS working solution (Elbasvir, 400 ng/mL). The mixture was vortexed for 30 seconds to ensure proper mixing.
Subsequently, 500 µL of diluent (ammonium formate buffer, pH 3.0) and 300 µL of acetonitrile were added to precipitate plasma proteins. The samples were vortexed for 2 minutes and centrifuged at 4000 rpm for 20 minutes at room temperature.
The clear supernatant was carefully transferred into LC vials, and 10 µL was injected into the LC–MS/MS system for analysis.
Calibration standards and QC samples were prepared and processed in the same manner as study samples to maintain consistency.
This protein precipitation approach provided efficient extraction recovery with minimal matrix interference and ensured reproducible quantification of SOF, VOX, VEL, and the IS.
Method Validation
The developed LC–MS/MS method was validated in accordance with key principles of ICH M10 and US FDA bioanalytical method validation guidelines applicable to preclinical studies.
The validation parameters evaluated included selectivity, carryover, linearity, sensitivity (LLOQ), accuracy, precision, recovery, matrix effect, dilution integrity, and stability.
Selectivity was assessed using six independent lots of blank rat plasma to evaluate potential endogenous interference at the retention times of analytes and IS. Carryover was evaluated by injecting blank samples immediately after the highest calibration standard (ULOQ) to ensure that residual analyte response did not exceed 20% of LLOQ for analytes and 5% for the IS. Calibration curves were constructed using at least six non-zero concentration levels, and linearity was assessed using weighted (1/× 2 ) regression of analyte-to-internal standard peak area ratios. Accuracy and precision were evaluated at four QC levels (LLOQ, LQC, MQC, HQC) using six replicates across intra- and inter-day runs. Acceptance criteria were ±15% for QC samples and ±20% for LLOQ. Recovery and matrix effects were assessed using post-extraction spiking methods, with IS normalization. Dilution integrity was evaluated by analyzing samples above the upper limit of quantification (ULOQ) after appropriate dilution with blank plasma. Stability studies included bench-top, autosampler, freeze–thaw, and long-term stability under defined conditions. Due to the preclinical scope of the study, evaluation of hemolyzed and lipemic matrices was not performed and is acknowledged as a limitation.
Pharmacokinetic Study Application
The animal study was conducted in accordance with CPCSEA guidelines and institutional ethical standards. Ethical approval was obtained from the Institutional Animal Ethics Committee (IAEC) (Approval No. IAEC/SOPS/04/2025; Registration No. 2325/PO/Re/2025/CCSEA). Female Sprague–Dawley rats (n = 6) were housed under controlled environmental conditions (temperature 22°C ± 2°C, relative humidity 55% ± 5%, 12 h light/dark cycle) with free access to standard laboratory diet and water. Animals were acclimatized prior to the study. Randomization was performed before dosing to minimize bias. A single oral dose of SOF (10 mg/kg), VOX (5 mg/kg), and VEL (5 mg/kg) was administered. Blood samples were collected at predefined time intervals via minimally invasive techniques to reduce animal distress. All procedures were conducted in accordance with ethical guidelines for animal handling and welfare.
Statistical Analysis
Calibration curves were generated using weighted (1/× 2 ) linear regression of analyte-to-internal standard peak area ratios. Pharmacokinetic parameters, including Cmax, Tmax, AUC₀-t, AUC₀-∞, and t½, were calculated using non-compartmental analysis (NCA) in Phoenix WinNonlin software. Data are presented as mean ± standard deviation (SD). Given the exploratory nature of this preclinical study and the repeated-measures design, no inferential statistical comparisons (e.g., ANOVA) were performed between analytes. Pharmacokinetic results are interpreted descriptively.
Results
Method Development
The method was developed to achieve optimal peak symmetry, resolution, and short retention times for SOF, VOX, and VEL. After several trials with varying mobile phase compositions, pH, and organic modifiers, the final optimized condition ensured optimal chromatographic performance and minimized carryover between injections.
During method development, different chromatographic conditions were evaluated by varying the column type and mobile phase composition to achieve optimal separation of SOF, VOX, VEL, and the IS Elbasvir. Initially, an X-Bridge C18 column with acetonitrile–0.1% perchloric acid mobile phase was tested, but inadequate plate count and peak tailing were observed. Further trials with acetonitrile–perchloric acid and acetonitrile–ammonium formate buffer systems showed either insufficient resolution or peak broadening. Finally, satisfactory chromatographic performance was obtained using an Agilent Eclipse XDB C18 column (250 × 4.6 mm, 5 µm) with acetonitrile and ammonium formate buffer (20:80, pH 3.0 adjusted with formic acid), which produced well-resolved peaks with acceptable system suitability parameters. The retention times were approximately 2.13 min for SOF, 3.29 min for VOX, 4.62 min for the IS (Elbasvir), and 5.56 min for VEL, confirming effective chromatographic separation without matrix interference.
The ionization and detection conditions for mass spectrometry were optimized to ensure sensitivity. The ESI⁺ mode with MRM transitions provided precise and reproducible detection. The optimized parameters yielded strong, stable signal intensities for all analytes with minimal noise and carryover effects. The mass spectrometer was operated using an electrospray ionization (ESI) interface in positive ion mode. Quantification of SOF, VOX, VEL, and the IS Elbasvir was performed using MRM mode. The optimized mass spectrometric parameters were as follows: ion spray voltage 5500 V, source temperature 550°C, collision energy 14 V, declustering potential 40 V, entrance potential 10 V, exit potential 7 V, and dwell time 1 s. Nitrogen was used as the collision gas, and the drying gas temperature was maintained between 120°C and 250°C with a flow rate of 5 mL/min. These optimized conditions provided stable ionization and sensitive detection for all analytes. The optimized method was found robust, with consistent peak responses and reliable detection at nanogram levels, suitable for pharmacokinetic and bioequivalence applications.
Method Validation
Selectivity and Specificity
Assessed using six different rat plasma lots, with no interfering peaks observed at the retention times of SOF, VOX, VEL, or the IS. Blank and LLOQ chromatograms showed clean, well-resolved peaks, confirming no endogenous interference. Specificity was evaluated in accordance with USFDA and ICH M10 bioanalytical validation guidelines by analyzing six different blank rat plasma lots to assess potential endogenous interference. Blank plasma samples, blank plasma spiked with IS, and plasma samples spiked at LLOQ levels were examined. No interfering peaks exceeding 20% of the LLOQ response for analytes or 5% for the IS were observed at their respective retention times, confirming adequate method specificity.
Carryover
Carryover was evaluated by injecting blank plasma samples immediately after the ULOQ. No significant carryover was observed, as analyte responses in blank samples were <20% of the LLOQ and <5% for the IS, meeting acceptable criteria.
Dilution Integrity
Dilution integrity was assessed by analyzing samples spiked above the ULOQ and diluted with blank plasma. The diluted samples showed acceptable accuracy (within ±15%) and precision (%CV ≤ 15%), confirming that samples exceeding the calibration range can be reliably quantified after dilution.
Linearity
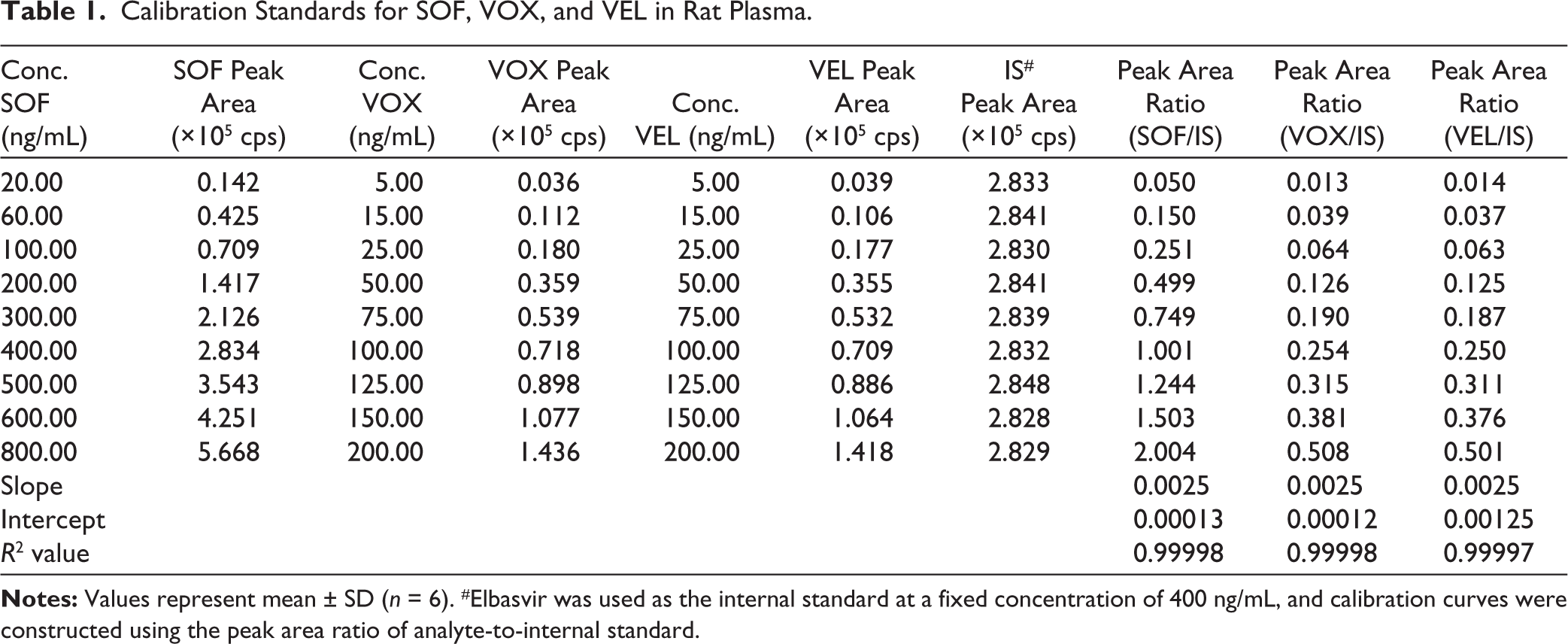
The selected calibration ranges were based on expected pharmacokinetic plasma concentrations following oral administration. Preliminary pilot studies and literature reports indicated that SOF plasma concentrations typically reach several hundred ng/mL after dosing, while VOX and VEL concentrations are comparatively lower. Therefore, calibration ranges of 20–800 ng/mL for SOF and 5–200 ng/mL for VOX and VEL were selected to adequately capture both peak and terminal phase concentrations. Established excellent linearity over 20–800 ng/mL for SOF, 5–200 ng/mL for VOX, and 5–200 ng/mL for VEL. Calibration curves used analyte/IS peak area ratios with 1/× 2 weighted linear regression (Figure 3). The correlation coefficients (R2) for all three analytes were consistently greater than 0.999, satisfying ICH M10 validation requirements (Table 1).
Linearity Plots of SOF, VOX, and VEL.

Calibration Standards for SOF, VOX, and VEL in Rat Plasma.
Sensitivity
The LLOQ was 20 ng/mL for SOF and 5 ng/mL for VOX and VEL, with acceptable precision and accuracy (%CV ≤ 2% and mean accuracy between 92% and 93%). The signal-to-noise ratios were more than tenfold at LLOQ levels, indicating excellent sensitivity. The achieved LLOQ values are adequate for preclinical pharmacokinetic applications and allow reliable quantification of analytes within the observed concentration range. However, the sensitivity is comparable to previously reported LC–MS/MS methods rather than superior, and this has been considered in the interpretation of results (Table 2).
Sensitivity Results (LLOQ) for SOF, VOX, and VEL.

Precision and Accuracy
The %CV values for SOF, VOX and VEL were found to be below 1.2%, 1.7%, and 1.9%, respectively, while inter-day %CV values remained below 2% for all analytes. Accuracy was determined by comparing the back-calculated concentration obtained from the calibration curve with the nominal (theoretical) concentration of the QC samples. Accuracy was expressed as a percentage and calculated using the following formula:

where the measured concentration represents the back-calculated value obtained from the calibration curve. Accuracy was evaluated at four QC levels (LLOQ, LQC, MQC, and HQC) using six replicates. The obtained accuracy values were within the acceptable limits specified by ICH M10 bioanalytical method validation guidelines (85%–115% for LQC, MQC, HQC and 80%–120% for LLOQ).
The mean accuracy ranged between 92% and 98%, demonstrating excellent repeatability and reproducibility, which confirms the methodology is precise and accurate. Accuracy (%) and recovery (%) were calculated using the following equations:

Recovery was evaluated by comparing peak areas of extracted QC samples with those of post-extraction spiked samples at equivalent concentrations (Tables 3 and 4).
Quality Control (QC) Samples for SOF, VOX, and VEL.

Precision and Accuracy for SOF, VOX, and VEL.

Recovery and Matrix Effect
Recovery of SOF, VOX and VEL, and IS ranging between 96% and 98% with %CV <0.5%. The results indicated negligible ion suppression or enhancement, with mean accuracies within 95%–98%, confirming minimal matrix interference and high extraction efficiency (Table 5). These findings validate the robustness of the sample preparation technique using acetonitrile-induced protein precipitation. Recovery and matrix effect evaluation were performed using IS–normalized responses (Figure 4).
Recovery and Matrix Effect for SOF, VOX, and VEL.

Chromatograms Representing Different Levels of Concentrations.

Stability Studies
Stability was evaluated at multiple QC levels (LQC, MQC, HQC) to ensure reliability across the calibration range. Long-term stability was assessed at multiple time points (Day 1, 7, 14, 21, and 28), providing a time-dependent stability profile of analytes under frozen storage conditions. Bench-top stability was evaluated at 25°C ± 2°C for 12 h, freeze–thaw stability after three cycles between −20°C and room temperature, autosampler stability at 10°C for 24 h, and long-term stability at −20°C for 28 days. Results in all studies with %CV values below 2% and mean accuracy within 89%–90% of nominal values. Long-term stability refers to the stability of analytes during prolonged storage of biological samples under frozen conditions prior to analysis (Tables 6 and 7). The analytes remained stable for at least 24 hours in the autosampler, three freeze–thaw cycles, and 12 hours under bench-top conditions, confirming that sample handling and processing do not affect quantification integrity.
Combined Stability Data for SOF, VOX, and VEL.
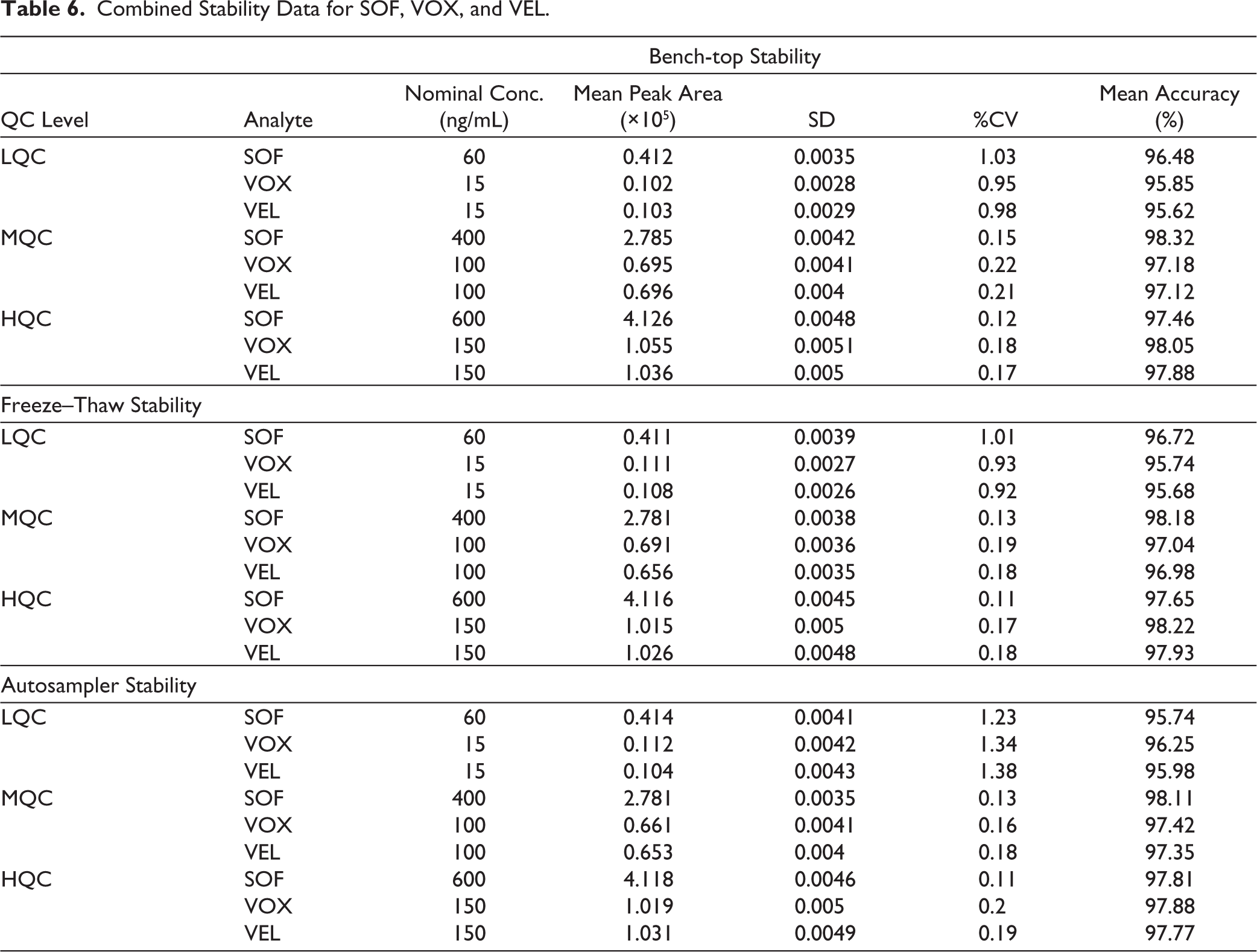
Combined Stability Data for SOF, VOX, and VEL.

Pharmacokinetic Application Results
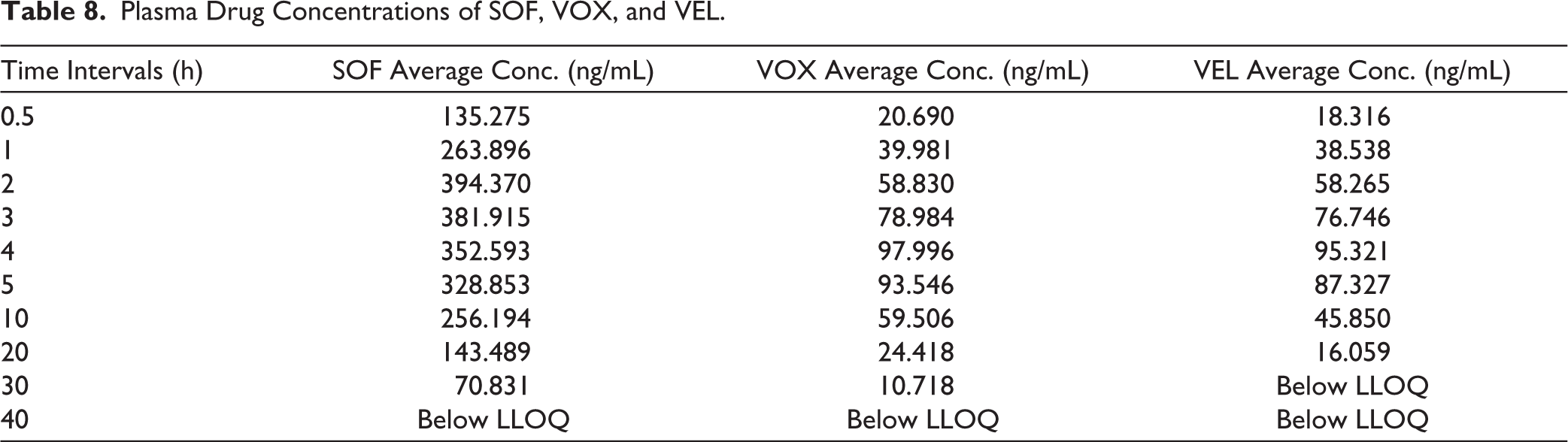
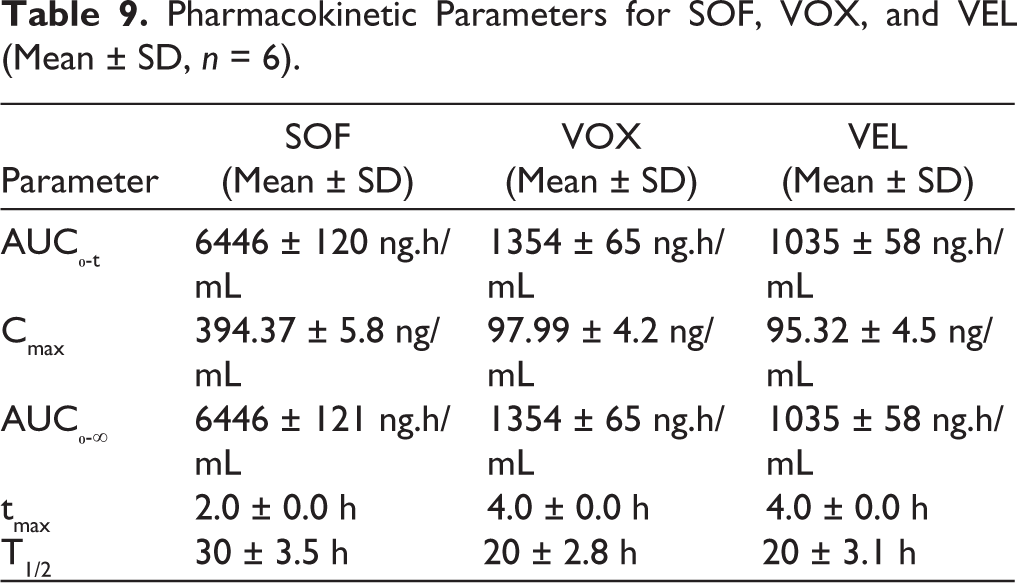
The validated LC–MS/MS method was successfully applied to the pharmacokinetic evaluation of SOF, VOX, and VEL following oral administration in female Sprague–Dawley rats. The plasma concentration–time profiles demonstrated distinct absorption characteristics for each analyte. SOF exhibited relatively rapid absorption, reaching peak plasma concentration (Cmax) at approximately 2 hours (Tmax), whereas VOX and VEL reached Cmax at approximately 4 hours, indicating comparatively slower absorption. The observed differences in Cmax values are consistent with the administered doses (SOF: 10 mg/kg; VOX and VEL: 5 mg/kg) and reflect dose-dependent systemic exposure. Systemic exposure, as assessed by AUC₀-t, followed the order SOF > VOX > VEL. AUC₀-∞ values were approximately equal to AUC₀-t due to negligible measurable concentrations beyond the final sampling point; however, this may reflect limitations in terminal phase characterization (Figure 5). Additionally, the number of sampling time points was limited, which may affect the accurate estimation of terminal pharmacokinetic parameters (Tables 8 and 9).
Mean Plasma Concentration-time Profiles of SOF, VOX, and VEL Following Oral Administration in Rats (mean ± SD, n = 6).

Plasma Drug Concentrations of SOF, VOX, and VEL.
Pharmacokinetic Parameters for SOF, VOX, and VEL (Mean ± SD, n = 6).
Comparison with Existing Methods
Several analytical methods have been reported for the determination of SOF, VEL, and VOX using HPLC, UPLC, and LC–MS/MS techniques. Many of these methods focus on individual analytes or binary combinations and are often developed for pharmaceutical formulations rather than biological matrices. LC–MS/MS methods reported in the literature have demonstrated comparable or, in some cases, lower limits of quantification, particularly for SOF and VEL in human plasma. Therefore, the sensitivity achieved in the present study is considered adequate for preclinical pharmacokinetic applications rather than superior. The primary contribution of the present work lies in the simultaneous quantification of SOF, VOX, and VEL in rat plasma using a straightforward protein precipitation technique and a relatively short run time. This approach provides a practical and reproducible analytical method suitable for exploratory pharmacokinetic studies.
Discussion
The results confirm that the developed LC–MS/MS method is reliable, accurate, and efficient for simultaneous quantification of SOF, VOX, and VEL in rat plasma. The optimized chromatographic conditions provided effective separation and sharp, well-defined peaks for all analytes and the IS. The optimized mass spectrometric parameters provided adequate sensitivity and resolution, enabling quantification at nanogram levels with minimal matrix interference.
The method’s validation, carried out according to ICH M10 bioanalytical guidelines, confirmed its robustness, precision, and reproducibility. Parameters such as selectivity, linearity, accuracy, precision, recovery, and stability met the defined acceptance criteria, ensuring that the method is suitable for reliable routine bioanalysis. The simplicity of the protein precipitation extraction procedure further enhances the method’s applicability, making it both time-efficient and cost-effective for high-throughput analysis in pharmacokinetic studies.
Application of the method to the pharmacokinetic study in rats demonstrated its suitability for in vivo analysis. The pharmacokinetic profiles indicated that SOF achieved higher systemic exposure compared with VOX and VEL, reflecting its distinct absorption and metabolic behavior. However, all three analytes exhibited similar elimination half-lives (t½), indicating comparable clearance kinetics. These results are in alignment with the known pharmacological properties of these DAAs and provide insight into their combined absorption and elimination characteristics.
By offering a validated LC–MS/MS method for simultaneous quantification of SOF, VOX and VEL, this study significantly enhances the analytical toolkit available for antiviral pharmacokinetic and bioequivalence research. Collectively, the findings confirm the method’s robustness and reliability, establishing it as a valuable analytical platform for combination antiviral therapy research. The developed LC–MS/MS method provides a robust analytical platform that can be extended to several future research directions. The method demonstrates suitability for preclinical pharmacokinetic investigations. Application to clinical pharmacokinetic or therapeutic monitoring studies requires further validation in human biological matrices in accordance with regulatory requirements. Furthermore, the analytical strategy could be adapted for the quantification of these antivirals and their metabolites in additional biological matrices such as human plasma, cerebrospinal fluid, or tissue samples. While the developed method demonstrates adequate performance for preclinical applications, its sensitivity is comparable to existing LC–MS/MS methods reported in the literature. Furthermore, the method has been validated only in rat plasma, and its applicability to human pharmacokinetic or clinical studies requires additional validation in human biological matrices in accordance with regulatory requirements.
Therefore, the present study should be considered as a preclinical analytical and pharmacokinetic investigation, and extrapolation to clinical settings should be approached with caution. The sampling duration may not fully characterize the terminal elimination phase, which may affect the accuracy of estimated pharmacokinetic parameters such as AUC₀-∞ and t½.
Limitations
The pharmacokinetic sampling duration presented in this study may not fully capture the terminal elimination phase, and the relatively sparse sampling design limits detailed pharmacokinetic interpretation. The bioanalytical method was validated only in rat plasma, and matrix variations such as hemolyzed and lipemic conditions were not evaluated. Although key validation parameters were assessed, full regulatory validation requirements for clinical studies were not addressed. Therefore, further studies are required to extend the applicability of this method to human biological matrices and clinical pharmacokinetic investigations.
Conclusion
A reliable and reproducible LC–MS/MS method was developed for the simultaneous quantification of SOF, VOX, and VEL in rat plasma. The method demonstrated acceptable accuracy, precision, recovery, and stability in accordance with key principles of bioanalytical method validation. The method was successfully applied to a preclinical pharmacokinetic study, demonstrating its suitability for exploratory in vivo analysis. However, the pharmacokinetic findings should be interpreted as preliminary due to limitations in sampling design. Overall, the developed method provides a practical analytical tool for preclinical pharmacokinetic investigations. Further studies are required to extend its applicability to human matrices and clinical pharmacokinetic evaluations.
Footnotes
Acknowledgements
The authors would like to thank Biocon Biologics Limited, Bengaluru, India, for providing gift samples of the analytes and internal standard. The authors also acknowledge the institutional facilities provided for carrying out this research work.
Authors’ Contribution
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Data Availability Statement
All the data is available with the authors and shall be provided upon request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval was obtained from the Institutional Animal Ethics Committee (IAEC) (Approval No. IAEC/SOPS/04/2025; Registration No. 2325/PO/Re/2025/CCSEA).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Not applicable.
Use of Artificial Intelligence-assisted Tools:
AI-based language assistance tools were used solely for improving the clarity of language, grammar, and overall readability of the manuscript. All such suggestions were carefully reviewed, critically evaluated, and edited by the authors to ensure accuracy, scientific integrity, and consistency with the original work. The authors take full responsibility for the content of the manuscript, and all revisions reflect the authors’ scientific judgment and expertise.