
Abstract
Background:
The phenotypic variability of C9orf72-associated disease is broadening, including atypical and non-motor presentations. C9orf72-associated neurodegeneration has only rarely been associated with primary lateral sclerosis (PLS), and even more rarely with ocular motor apraxia.
Objectives:
Describe a family with C9orf72 mutation presenting with frontotemporal dementia (FTD) and atypical PLS phenotypes and discuss the implications regarding 1) where PLS lies on the ALS-FTD spectrum, and 2) how C9orf72 mutations influence PLS clinically.
Methods:
Chart review.
Results:
A 52-year-old male experiencing 4 months of progressive right lower leg spasticity with a family history of FTD was referred to us. Within 15 months, he was anarthric and required a powered wheelchair. He developed acquired ocular motor apraxia, consistent with supranuclear ophthalmoplegia. He later developed laryngeal dystonia which led to his death. Ten years later, his 67-year-old brother presented with 8 months of progressive spastic dysarthria, hyperreflexia, right foot drop, and right facial weakness. Genetic testing revealed heterozygous C9orf72 hexanucleotide repeat expansion.
Conclusions:
This family's presentation expands on sparse reports of C9orf72-associated PLS. The proband showcases a severity of ocular motor deficits not yet reported in PLS, extending ocular motor findings in MND. These deficits also provide clinical evidence of degeneration outside the motor cortex/spinal cord in PLS. The symptomatology (laryngeal dystonia, rapid progression) clinically overlaps with ALS/FTD, suggesting PLS may lie on the ALS-FTD spectrum. The severity and atypicality of this case also support suggestions that C9orf72 mutations amplify the spectrum/severity of disease observed in TDP-43 proteinopathies.
Keywords
Introduction
C9orf72 hexanucleotide repeat expansions are responsible for 40–50% of familial amyotrophic lateral sclerosis (ALS) cases and up to 10% of sporadic cases. 1 These mutations are also observed in nearly 25% of familial frontotemporal dementia (FTD) cases. 2 Primary lateral sclerosis (PLS) is a restricted upper motor neuron (UMN) form of ALS and has been defined as the absence of significant active lower motor neuron (LMN) degeneration four or more years after symptom onset. 3 PLS has also been associated with C9orf72 mutations, albeit at a greatly reduced rate compared to ALS and FTD (i.e., 1.6%). 4
Mutations at the C9orf72 locus are associated with a broad spectrum of phenotypes. This spectrum continues to expand, and new phenotypes associated with C9orf72 mutations may provide insight into the diverse pathogenic effects of the mutation. Recently, higher-order oculomotor abnormalities have been observed in presymptomatic patients with C9orf72 mutations. 5 Similar higher-order oculomotor abnormalities have been observed in FTD, 6 and gaze palsies have been observed in late-stage ALS (without confirmed C9orf72). 7 However, while mounting evidence suggests the presence of executive ocular motor deficits in PLS,8–10 and some studies describe reduced range/velocity of extraocular movements in particular directions 11 (including one report of vertical supranuclear gaze palsy 12 ), there have been no documented cases of complete loss of volitional eye movements in PLS. We therefore describe this family to expand on the range of phenotypes observed in PLS and in C9orf72-mediated motor neuron disease (MND) more broadly. We then use our cases to spur a review of literature pertaining to C9orf72-mediated MND, specifically a hypothesis pertaining to TDP-43 proteinopathies.
Methods
With written consent, we performed a chart review of hospital records to describe two brothers: one diagnosed with definite PLS who developed an acquired ocular motor apraxia, and the other diagnosed with probable PLS. Their family history was notable for FTD in their maternal aunt. The living subjects of the described family provided written informed consent for publication. Review by our institutional ethics review board was not required.
Case description
Case 1
A 52-year-old male patient was referred to our neuromuscular clinic with a 4-month history of progressive right lower limb spasticity. Three months after symptom onset (month 3), he required a cane. Muscular strength/bulk were preserved, however hyperreflexia was noted in all limbs, particularly on the right side where ankle clonus and an equivocal Babinski sign were observed. Cranial nerve (CN) exam revealed mild UMN-pattern right facial weakness and left-sided CN4 palsy felt to be of congenital origin. Pertinent negatives included sensory and autonomic disturbances. Laboratory tests (i.e., Lyme serology, B12, TSH, CK, Troponin I [< 0.02 µg/L]) were unremarkable aside from positive anti-dsDNA in the setting of negative ANA. Past medical history included livedo reticularis and a remote history of sarcoidosis (25 years prior). These findings prompted a chest CT (negative) and a trial of prednisone which did not abate symptoms. MRI scans were unremarkable aside from mild lumbar (L4/L5) foraminal stenosis. Genetic testing was negative for adult polyglucosan body disease; additional testing was declined. The patient's family history was remarkable for FTD in a maternal aunt (diagnosed ∼50 years old; unsure if autopsy performed, likely clinical diagnosis), and dementia featuring disinhibition and Parkinsonian features in his mother and paternal grandmother, respectively.
Electrodiagnostic studies performed externally in month 3 were normal. Upon presenting to our clinic in month 4, repeat needle electromyography (EMG) demonstrated sparse fibrillation potentials and positive sharp waves in the right medial gastrocnemius (S1 myotome) and right tibialis anterior (L4/L5 myotome) muscles. These findings were attributed to the aforementioned L4/L5 lumbar foraminal stenosis. Needle EMG findings were otherwise normal in the right rectus femoris, left lower extremity (L3-S1 myotomes), bilateral upper extremities (right infraspinatus, right deltoid, left extensor digitorum communis, left triceps), and axial muscles (right T8 thoracic paraspinal muscles). Nerve conduction studies were normal. Given the regionally confined abnormalities with alternative explanation, and absence of hallmark EMG signs suggesting LMN involvement, these results did not meet formal El Escorial criteria for ALS. 13
Within one year of symptom onset, there was considerable clinical decline. Cranial nerve exam was notable for worsening right lower facial weakness in an UMN pattern, spastic dysarthria, and reduced gag reflex. There was worsening spasticity and hyperreflexia in all four limbs with clonus and extensor plantar responses bilaterally. Follow-up brain MRI showed hyperintensity of the corticospinal tract. Baclofen and subsequently dantrolene were used for control of spasticity; anticholinergics, suctioning, and Botox were used to control secretions. The deterioration confined the patient to a wheelchair and necessitated a personal support worker for activities of daily living (ADLs).
By month 16, the patient was anarthric and communicated through an assistive device. Despite this clinical progression and increasing UMN symptoms, repeat EMG and nerve conduction studies revealed no progression of findings from month 4. This strengthened the possibility that these deficits were related to the patient's degenerative disc disease (L4/L5 foraminal stenosis). However, in the left arm, hand, and tongue, we observed decreased activation in an UMN pattern. Similar findings were replicated in needle electrode examination of the upper and lower extremities in month 19 and month 33. Pulmonary function tests (PFTs) at this time revealed a forced vital capacity (FVC) of 58% predicted confirming respiratory muscle weakness. The patient's significant clinical deterioration without clinical or EMG evidence of progressive LMN involvement most supported a diagnosis of PLS according to the most recent consensus diagnostic criteria. 3 UMN-dominant ALS was initially considered per previous diagnostic criteria, 14 but felt to be less likely.
In month 16, Riluzole was started. By month 17, the patient developed a rightward horizontal gaze palsy whereby he was unable to voluntarily move either of his eyes rightwards. Ophthalmology was consulted at this time. Their examination revealed that volitional saccadic and smooth pursuit eye movements were diminished in all directions but particularly horizontally. These limitations were overcome with oculocephalic/optokinetic manoeuvres, and when saccades were reflexively evoked to audiovisual stimuli. These findings constituted an acquired ocular motor apraxia. His congenital left CN4 palsy worsened, causing left hypertropia and resulting diplopia. The diplopia was ameliorated using prism glasses. Later, in month 19, vertical eye movements became significantly more limited, and the patient had essentially no volitional eye movements. In month 21, he began experiencing blurred vision. This was thought to be due to the lack of ocular movements, as these findings were less severe for smaller and more distant objects which required less re-foveation. In month 29, the patient's visual acuity in the right eye suddenly worsened (OD: 20/60, OS: 20/40). Fundoscopic exam was normal.
Altogether, the ocular findings were interpreted to reflect severe supranuclear deficits causing acquired ocular motor apraxia (potential degeneration of frontal/parietal eye fields, striata) and mild infranuclear involvement due to the congenital CN4 palsy. The severity of this oculomotor abnormality fluctuated mildly throughout the disease course, as did the direction of gaze which was predominantly affected (i.e., horizontal vs. vertical).
The patient experienced three generalized tonic-clonic seizures in one day in month 23, which prompted presentation to the emergency room where seizures abated with administration of Ativan. The seizures were thought to be provoked either by the patient's three-day history of benzodiazepine withdrawal or vomiting-induced hyponatremia. Lumbar puncture was unremarkable. EEG revealed potentially epileptiform discharges in the right posterior quadrant, exacerbated by drowsiness/slow-wave sleep. Over the next year, he experienced episodes suggestive of absence seizures, however repeat EEG (month 35 and 37) revealed no overt seizure activity. By month 33, he could no longer produce any volitional limb movements. In month 77, the patient's wife began observing intermittent but progressively intensifying inspiratory stridor and low-pitched upper airway sounds. Weeks later, consultation with ENT with direct nasolaryngoscopy revealed nearly completely adducted vocal cords with no abduction on inspiration, consistent with laryngeal dystonia. Options were presented for the treatment of laryngeal dystonia, but unfortunately, the patient died in month 79. Altogether, given the clinical and EMG evidence of diffuse UMN involvement without LMN involvement for a duration of greater than four years, diagnostic criteria for definite PLS are fulfilled in this patient. 3
Case 2
Ten years later, the proband's brother (67 years old) presented to our neuromuscular centre in with an 8-month history of progressive bulbar dysfunction characterised by spastic dysarthria, dysphagia, and drooling. Neurological exam revealed no limb abnormalities, and medical history was unremarkable in the context of these symptoms. MUSK and ACh-R antibodies tests were negative, and there were no signs of REM sleep behaviour disorder. Brain MRI performed 7 months after symptom onset (month 7; prior to presentation to our clinic) revealed a small-to-2 mm focus of T2 FLAIR hyperintensity in the left precentral gyrus along the anterolateral hand knob. Overall, given his lack of limb abnormalities on neurological examination, this was felt to be an incidental finding of remote ischemia with no clinical correlate. Considering the patient's family history, symptoms were interpreted as suspicious for bulbar-onset motor neuron disease.
By month 11, his speech was significantly more impaired, and dysphagia had become present with both thick solids and thin liquids. He developed mild bifacial weakness and right-sided ptosis. He was also increasingly emotional, suggesting pseudobulbar affect. By this time, patellar reflexes were very brisk (3+) and there was evidence of spasticity. Despite worsening clinical features, EMG studies were entirely normal. Genetic testing using two repeat-primed PCR assays 15 and two fluorescent fragment-length assays 16 confirmed a heterozygous C9orf72 hexanucleotide repeat expansion; unfortunately, the full ALS sequencing panel and ATXN2 testing were therefore waived per company protocol (Prevention Genetics, USA). The tests used by Prevention Genetics are qualitative in nature and not designed to quantify the number of C9orf72 repeats; however, the company regards repeat lengths of < 30 as normal (and their test methodology 15 is validated for reliably detecting the presence/absence of expansions greater than ∼100 repeats).
The patient remained active, ambulating independently and working full-time. However, by month 14, he reported a right foot drop and 5-pound weight loss. Strength of his neck extensors and flexors, and hip abductors, were slightly decreased (4+/5) and his right ankle dorsiflexion was moderately weak (4/5). Treatments with Riluzole and Edaravone were initiated.
By month 17, the FVC was 68% but due to impairment of inspiratory and expiratory pressures, nightly bilevel ventilation (BiPAP) was initiated. The patient began experiencing a severe head drop requiring a soft brace and reduced tongue movements without fasciculations. On most recent assessment in month 29, his dysarthria had progressed such that he was essentially unable to speak. His neurological status was otherwise unchanged. Notably, there remained no clinical evidence of LMN involvement. As such, given the clinical evidence of UMN involvement, and the lack of clinical/EMG evidence of LMN involvement after 29 months of disease, diagnostic criteria for probable PLS are fulfilled in this patient. 3
Discussion & conclusions
This family's presentation of definite PLS with severe supranuclear ocular motor deficits, probable PLS in a sibling, and FTD in the maternal aunt expands on the spectrum of clinical findings observed in PLS. The severity of the ocular motor deficits is novel to both PLS and patients and C9orf72-mediated disease more broadly. The symptomology of the case of PLS also overlaps with ALS, featuring laryngeal dystonia and an atypically aggressive course.
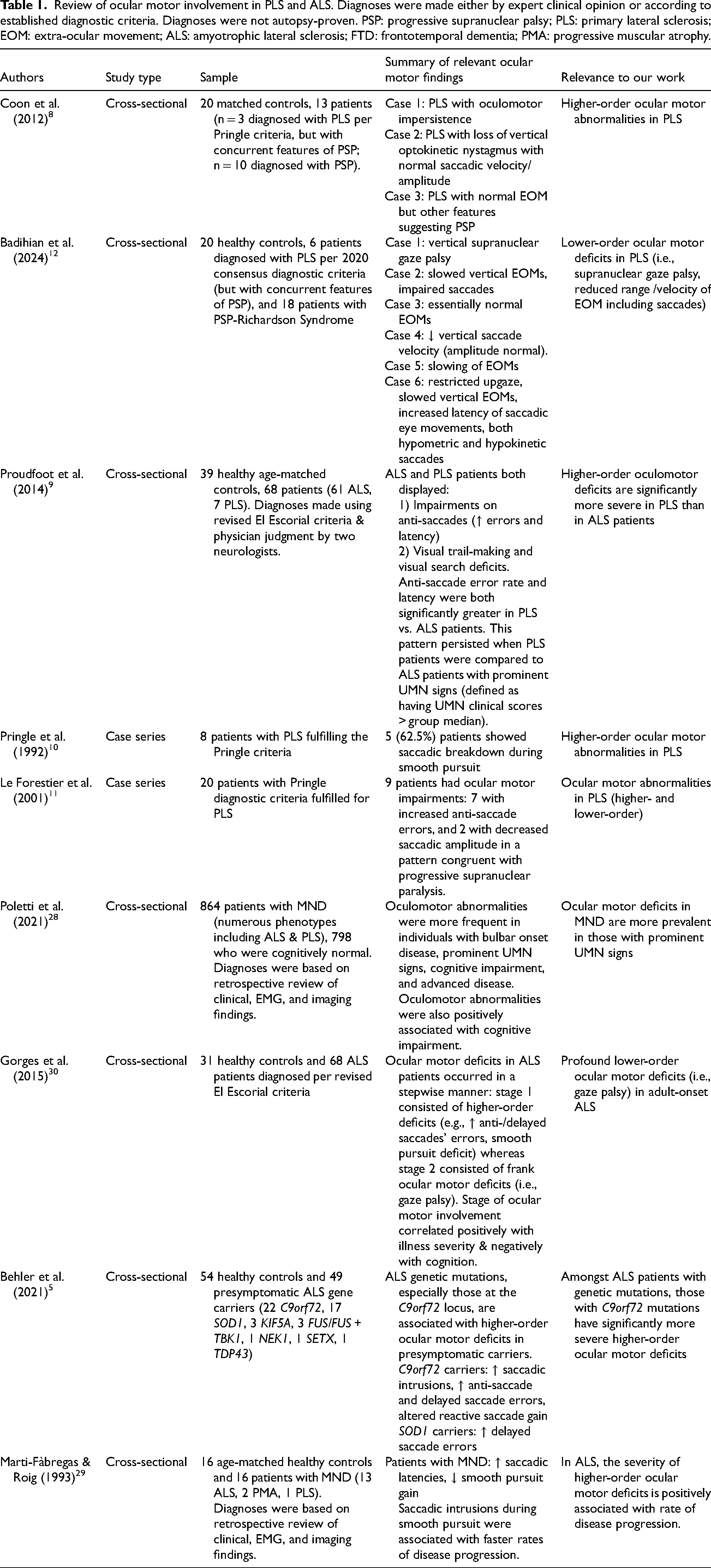
The proband's acquired ocular motor apraxia aligns with neuroimaging17,18 and clinical19–21 evidence that neurons outside the motor cortex/spinal cord are degraded in PLS, parallelling ALS research showing extramotor degeneration along frontotemporal and extrapyramidal axes (for review, see 22 ). This apraxia likely reflects bilateral degeneration of frontal/parietal eye fields23–25 or striata, 26 sparing the downstream lower motor neurons and thus explaining the preserved reflexive but absent voluntary eye movements. This ocular motor apraxia draws parallels to the clinical and degenerative pattern typically observed in PLS; in both ocular motor apraxia and PLS, the lower-level neurons directly responsible for task execution are spared, while higher-order neurons are lost. The deficits associated with these conditions reflect this (i.e., often failing to reach the level of total plegia). Altogether, our case provides evidence of probable degeneration outside of the primary motor cortex/spinal cord and expands upon the existing descriptions of ocular motor deficits in PLS. For review of the ocular motor deficits in PLS (and MND more broadly) which are pertinent to our case, see Table 1. Most notably, higher-order (executive) oculomotor deficits have been extensively implicated in ALS 27 and MND, particularly in those with prominent UMN signs 28 and rapidly progressive disease. 29 Proudfoot et al. (2014) showed that these higher-order deficits are more severe in PLS compared to ALS. 9 Other works have also described ocular motor deficits in PLS,8,10–12 though none as severe as those observed in our case. Notably, given our case's particularly aggressive disease course and extent of ocular motor deficits, there is evidence that ocular motor deficits in ALS progress in a stepwise manner from higher- to lower-order in parallel with global disease progression. 30
Review of ocular motor involvement in PLS and ALS. Diagnoses were made either by expert clinical opinion or according to established diagnostic criteria. Diagnoses were not autopsy-proven. PSP: progressive supranuclear palsy; PLS: primary lateral sclerosis; EOM: extra-ocular movement; ALS: amyotrophic lateral sclerosis; FTD: frontotemporal dementia; PMA: progressive muscular atrophy.
TDP-43 mislocalization from nucleus to cytoplasm is a pathologic hallmark in both ALS31,32 and PLS. 33 Given this, the presence of C9orf72 34 hexanucleotide repeat expansions (which induce TDP-43 proteinopathy 35 ) in both ALS36,37 and PLS 4 is unsurprising. Thus, while no autopsy was performed on our proband, his diagnosis of PLS and likely C9orf72 mutation suggest that his disease is a TDP-43 proteinopathy. We do note, however, that we cannot be certain without autopsy evidence. Regarding C9orf72 mutations, the phenotypic variability associated with these mutations is broad and expanding, including recent works outlining associations with Huntington-like disorders, corticobasal syndrome, Lewy body dementia, 38 and psychiatric illness. 39
Despite this growing phenotypic variability, the severity of oculomotor deficits in our patient has not, to our knowledge, been reported previously in C9orf72 patients. Interestingly, however, in C9 vs. wild-type MND patients, hypometabolism/degeneration has been observed40,41 in regions which may underlie ocular motor apraxia and explain our patient's presentation (e.g., frontal and parietal eye fields,23–25 striata 26 ). To our knowledge, this case provides the first behavioural findings in accordance with such C9orf72-associated dysfunction/degeneration.
The proband's atypically severe features (e.g., laryngeal dystonia, 42 rapid progression, 43 profound oculomotor deficits 7 ) are not typically associated with PLS, and overlap with ALS symptomatology. This is in concordance with recent work suggesting PLS lies on the ALS-FTD spectrum of disease.44–46 Finally, given that C9orf72 mutations are associated with a significantly earlier age of onset and shortened survival amongst ALS patients,1,47 a similar pattern may exist in PLS and would explain our patient's particularly rapid and severe progression.
Compounding this genotype-phenotype interaction, the clinical features expressed in C9orf72-mediated disease may be further influenced by interactions with other concurrent genetic variants (“genetic modifiers”). 48 In this regard, it is possible that the relative sparing of LMNs in PLS is related to genetic modifiers in these patients which render their LMNs resistant to TDP-43-mediated degeneration. Indeed, recent work suggests that expression of certain inhibitory presynaptic proteins is protective to LMNs against TDP-43-mediated degeneration. 49 If this were the case, and both a pathogenic and protective genetic variant were therefore required for UMN-restricted phenotypes, then statistically it is unsurprising that (1) ALS and PLS co-occur within pedigrees50,51 and (2) PLS displays less consistent familial inheritance than ALS. 4
Our case of PLS (a TDP-43 proteinopathy) harbouring progressive supranuclear palsy-like (PSP; a tauopathy) gaze abnormalities is notable as the reverse has also been observed (i.e., autopsy-confirmed tauopathy presenting as PLS without any gaze palsy). 52 PLS-like syndromes have also been associated with Presenilin-1 mutations. 53 These findings underscore the degree of potential phenotypic overlap between distinct proteinopathies, highlighting the need for caution when inferring TDP-43 pathology without autopsy confirmation. This phenotypic overlap between proteinopathies may also reflect shared high-level factors which can selectively localize pathologic proteins of numerous kinds specifically to UMNs. While these potential high-level factors have yet to be elucidated, this aligns with recent work revealing significant overlaps in the upstream pathological cascades driving tauopathies and TDP-43 proteinopathies,54,55 and work demonstrating concurrent 56 and synergistic55,57,58 effects of TDP-43 and tau pathologies on one another in FTD and MND. Aligning with this, concurrent clinical and autopsy-proven ALS and PSP has been observed. 59 As such, our proband's symptoms may reflect the co-existence of TDP-43 and tau pathology, with one pathologic protein seeding and potentiating the effects of the other (and thus expressing a mixed phenotype with characteristics of both TDP-43- and tau-mediated disease).
Given the rarity of PLS, the current paper contributes to the limited existing literature pertaining to this rare but debilitating disease for which disease-modifying therapeutics are unavailable. We describe a family presenting with FTD, probable PLS, and definite PLS phenotypes. The case of definite PLS features a variety of highly atypical and severe clinical characteristics, the extent of which have not yet been observed in PLS. We hope that this will further elaborate on the phenotypic variation which may be observed in PLS and C9orf72-mediated diseases, expanding clinicians’ suspicions for this disease and mutation in patients presenting with a variety of symptoms. Finally, by describing a novel phenotype and therefore a novel pathogenic effect of C9orf72 mutations, we hope to increase the understanding of this mutation.
Footnotes
Ethical considerations
Review by our institutional ethics review board was not required.
Consent to participate
Case description performed with written informed consent.
Consent for publication
Case description performed with written informed consent for publication.
Author contributions
All authors contributed to the conceptualization, methodology, interpretation, and writing (drafting, critical reviews, and editing) of this work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Ari Breiner is an editorial board member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer review.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during this study.