
Abstract
Background
Spinal muscular atrophy (SMA) is a severe neurodegenerative disease affecting children. Three innovative disease-modifying therapies (DMTs)—nusinersen, risdiplam, and onasemnogene abeparvovec—are available for treatment.
Objective
To provide a descriptive overview of patients enrolled in the Registre SMA France until July 22, 2024.
Methods
Registre SMA France is a multicenter, national observational registry that includes patients with SMA—children and adults, treated or untreated. Data collection began retrospectively in 2016 and prospectively in 2020, with a 10-year follow-up plan. The coordinating center is the neuropediatric department of Garches Hospital (AP-HP), while methodological and, regulatory and operational management, are provided by the Clinical Research Unit of AP-HP Paris-Saclay. Financial support is provided through unrestricted grants from Biogen, Novartis, and Roche. Data on patient characteristics, medical and surgical follow-up, treatments, adverse events, and quality of life are recorded via structured forms, with additional modules developed as required (e.g., hematological monitoring post-gene therapy in 2021). Data quality is ensured through routine checks and periodic monitoring.
Results
By July 22, 2024, 1299 patients from 59 centers were enrolled (299 SMA1, 502 SMA2, 469 SMA3, 19 SMA4, 10 presymptomatic). Of these, 76.2% received DMT (nusinersen: 46.1%, risdiplam: 23.2%, onasemnogene abeparvovec: 9.2%), with 21.5% undergoing sequential or combination therapy. Major complications included ventilatory support (SMA1: 69.9%, SMA2: 64.5%, SMA3: 18.1%), enteral feeding (SMA1: 56.2%SMA1), and spine surgery (SMA2: 24.5%). Survival was significantly higher in treated SMA1 and SMA2 cases.
Conclusion
This registry serves as a key resource for understanding the clinical course and treatment outcomes of SMA in the real world, supporting future research and informing clinical and policy decisions in the era of DMTs.
Trial registration
NCT04177134.
Introduction
Spinal muscular atrophy (SMA), an autosomal recessive neuromuscular disease, is characterized by motor neuron degeneration in the anterior horn of the spinal cord, resulting in progressive muscle weakness and paralysis.1–3 SMA is classified into several types based on the age of onset and motor function. The most severe and common type—infantile-onset SMA (type I or SMA1)- manifests initial symptoms between birth and 6 months of age. Conversely, later-onset SMA types II (SMA2) and III (SMA3) affect children at the age of 7–18 months and >18 months, respectively. Additionally, two rarer forms exist: the congenital form (SMA0) characterized by generalized paralysis and respiratory failure at birth, and the adult-onset form (type IV or SMA4). 4 SMA1 is the leading genetic cause of infant mortality due to progressive respiratory and bulbar muscle weakness and is the second most common fatal autosomal recessive disorder after cystic fibrosis. 5 More than 95% of SMA cases are genetically characterized by homozygous deletions of exon 7 in the survival motor neuron 1 (SMN1) gene, identified as the causal gene in 1995. 6 A smaller proportion (3–5%) of affected individuals carry a compound heterozygous deletion and pathogenic variant. Disease severity is modulated primarily by the number of SMN2 gene copies, which encode a less stable SMN protein variant.7–10 Due to poor prognosis and early mortality in patients with SMA1 prior to the era of effective therapies, SMA2 and SMA3 became relatively more prevalent in the population.
The estimated incidence of SMA is 1 in 6000 to 1 in 10,000 live births, with a carrier frequency between 1/40 and 1/60. 11
Given its multisystemic impact, SMA management requires a multidisciplinary approach to address complex medical complications, including pulmonary, gastroenterological/nutritional, orthopedic assessments, and interventions by specialized teams under the supervision of experts in neuromuscular disorders, particularly SMA.12,13 Respiratory complications are a major cause of morbidity among patients with SMA1 and SMA2 and can also occur in a smaller proportion of patients with SMA3 following loss of ambulation. Owing to the degenerative nature of the disease, palliative and supportive care plays a critical role in patient management. 13
Major therapeutic breakthroughs over the past decade have dramatically changed the prognosis of SMA. The introduction of three disease-modifying therapies (DMTs)—nusinersen (SPINRAZA®), risdiplam (EVRYSDI®), and onasemnogene abeparvovec (OA, ZOLGENSMA®)—has significantly improved survival and motor function in affected individuals. 14 However, the high costs of these therapies limit their access worldwide. 15 Nusinersen, the first drug approved by the Food and Drug Administration and European Medicines Agency in 2016 for SMA, 16 is an antisense oligonucleotide administered intrathecally to enhance SMN protein expression via modulation of SMN2 splicing. Risdiplam, an orally administered splicing modifier, has been available in France since 2020. 17 OA, a gene therapy targeting SMN1 deficiency, became available in France in 2019 and has demonstrated efficacy in pre-symptomatic and symptomatic infants.18–22 Given the progressive nature of SMA, early treatment is critical, as irreversible motor neuron loss limits therapeutic benefits over time. Newborn screening programs for SMA have been implemented in different countries and regions worldwide. While universal newborn screening for SMA was not yet in place in France during the initial data collection period for this registry, a pilot program was underway in select regions, with nationwide implementation planned for 2025.23–27
Following the approval of these new treatments, the French Health Authority Haute Autorité de Santé [https://www.has-sante.fr/jcms/c_2822094/en/commission-de-la-transparence-reunion-du-31-janvier-2018] recommended systematic long-term, real-life data collection. In response, the Registre SMA France was established in January 2020, with academic coordination by the neuropediatric department of Garches Hospital (Assistance Publique–Hôpitaux de Paris, AP-HP), and methodological, regulatory, and operational oversight ensured by the Clinical Research Unit of AP-HP Paris-Saclay. The registry is supported by unrestricted grants from pharmaceutical stakeholders (Biogen, Novartis, and Roche). The Registre SMA France aims to document long-term, real-world evidence on SMA epidemiology, clinical progression, treatment efficacy, and safety outcomes. It includes both treated and untreated patients across all SMA types, allowing evaluation of various DMTs in real-life settings. To ensure adaptability, the registry includes specific modules that address emerging clinical needs. For example, a biological module was implemented in 2021 for infants receiving gene therapy, and a cognitive and neurodevelopmental module is currently being developed for SMA1-treated children.
This article does not aim to provide an exhaustive analysis of registry endpoints. Instead, it presents the structure, objectives, and initial descriptive insights of the Registre SMA France. Through a comprehensive overview of the enrolled population, collected data, and methodologies used, it illustrates the registry's potential as a research platform. Additionally, to ensure consistency with previous studies, we present key descriptive results and survival rate curves based on data collected up to July 22, 2024.
Methods and analysis
Registry design and setting
The Registre SMA France (NCT04177134) is a multicenter national observational registry 28 that enrolls all patients with genetically confirmed SMA1, SMA2, SMA3, or SMA4 who have been followed at participating centers since September 1, 2016. The registry includes pediatric and adult patients treated with DMT or not. Patients with SMA4 present with disease symptoms at ≥ 18 years of age. Congenital SMA, referred to as SMA0 by some authors, is categorized under SMA1. Data collection comprises both historical and prospective clinical parameters and follow-up data, including mortality, physical measurements, and cardiorespiratory, nutritional, and neuroorthopedic complications. Considering the significant impact of clinical care on disease progression, the registry meticulously documents medical interventions for each patient, including bracing, orthoses, surgeries, respiratory and feeding support, and rehabilitation therapies.
Outcome measures and parameters relevant to SMA and used in trials at French neuromuscular centers were collected to evaluate therapeutic efficacy and tolerability in real-life settings for the target population. Data were sourced from medical records, examination reports, complementary test results, measurements, and questionnaires completed by patients, their parents, or caregivers (for children) during routine follow-ups. Data collection is expected to span at least 10 years and be archived for 30 years. This data includes participants in three sub-cohorts: 1) historical sub-cohort of patients diagnosed or seen between September 1, 2016, and the center's participation date who were deceased or lost to follow-up on the day of or at least 1 day after the center's participation date; 2) historical-prospective sub-cohort of patients diagnosed or seen between September 1, 2016, and the center's participation date who were followed up on the day of or at least 1 day after the center's participation date; 3) prospective sub-cohort of patients diagnosed on or at least 1 day after the center's participation date, applicable only to newly diagnosed (incident) patients.
Eligibility criteria
All patients with genetically confirmed 5q SMA followed up at a French neuromuscular center (http://filnemus.fr) from September 1, 2016, were eligible. Genetic criteria include defects in both SMN1 genes, either due to homozygous deletion of exon 7 (0 SMN1), heterozygous deletion and a pathogenic variant (1 SMN1), or pathogenic variants in both SMN1 alleles (2 SMN1). Patients may be symptomatic (SMA1, SMA2, SMA3, and SMA4) or presymptomatic at the time of inclusion.
Ethics and dissemination
The registry protocol was approved by the Institutional Ethical Review Board of AP-HP, which was also responsible for data processing (protocol number: APHO200085). Participants (either parents or legal caregivers, in the case of minors) provided signed informed consent for their health data to be collected in the registry (full or non-identifiable partial data). Additionally, the enrollment procedure complied with the rules of the European Union's General Data Protection Regulation. No additional medical procedures outside standard SMA care are included. The results from this registry—manuscripts and other data reports (posters and communications at scientific conferences)—if submitted for publication in peer-reviewed journals and scientific forums, may be reviewed by the pharmaceutical companies financing the registry; however they cannot influence the results. Dissemination is performed through scientific forums and journals within national and international neuromuscular networks and by healthcare providers. Public summaries and monthly newsletters are available on the French Neuromuscular Network website (https://www.filnemus.fr/les-evenements-filnemus/actualites/).
Participants and sample size
Currently, the prevalence, incidence, and follow-up rates of SMA in France are unknown. Based on the number of patients reported by the neuromuscular center network in a previous national survey 29 and those identified by the French Administrative Healthcare Database (Système National des Données de Santé), the estimated number of patients with SMA of any type followed in these centers in 2019 was approximately 1000–1200 patients with SMA of any type (SMA1, SMA2, SMA3, and SMA4), half of whom were children. Available data from the coordinating center (Raymond Poincaré Hospital, Garches) revealed an SMA population (children and adults) of approximately 200 patients, accounting for approximately 20% of the national SMA population, with approximately 800–1000 patients estimated to be followed up across other participating centers.
Several American studies summarized by Lally et al. 30 estimated the birth prevalence of the SMA genotype at 8.5 to 10.3 per 100,000 live births in the USA. Of the 5q genotypes at birth, US studies have estimated the proportions of SMA1 (occurring in the first year), SMA2, and SMA3 to be 58%, 29%, and 13%, respectively, with the proportion of individuals with SMA4 estimated to be very low (∼1%). 11
SMA prevalence depends on medical practices and the availability of innovative treatments, which significantly alter the life expectancy of patients with early-onset, severe forms. In the pre-therapeutic era, survival primarily improved for patients with intermediate forms, attributed to advancements in ventilatory support, nutritional management, and surgical interventions to correct and stabilize spinal deformities developed in the 1970s and 1980s. In France, a progressive consensus emerged in the 1990s to adopt non-interventional management for infants with the more severe and common form (SMA0–1). In the absence of specific treatments, these children typically received palliative care and died early. Invasive respiratory support was deemed unreasonable for these infants owing to their extremely severe end-stage generalized paralysis, including facial and bulbar dysfunctions (locked-in syndrome). Consequently, early mortality in these cases contributed to a very low overall SMA prevalence. However, the introduction of new treatments available in France—nusinersen (2016), OA (2019), and risdiplam (2020)—has increased SMA prevalence. The nationwide implementation of newborn screening for the disease by 2025 is expected to further influence the overall prevalence of the affected population.
Objectives and endpoints
The primary objective of this real-life observational data collection is to provide a descriptive and dynamic analysis of the clinical characteristics, multisystemic functions, measurements, and health parameters of all children and adults with different SMA phenotypes, with a focus on the effectiveness and safety of the different therapies and supportive interventions used in France. Specific study objectives were defined based on the data collected, its completeness and quality (eTable 1). This article provides a descriptive global view of the enrolled population until July 22, 2024. Data entered after this date were not included.
Data monitoring
Automated procedures to identify missing or inconsistent data and outliers were developed by the AP-HP Clinical Research Unit. These data, known as data clarification forms, are communicated by Clinical Research Assistants (CRAs) to participating centers monthly, using Excel files for verification and correction. Data quality checks are also conducted via monitoring by CRAs using patient files in the centers and through weekly meetings with curators, CRAs, data managers, statisticians, and project managers.
Statistical analysis
The registry serves as a comprehensive research database which may be used in whole or in part depending on the objectives of each project. All scientific studies based on the registry must be supported by a detailed statistical analysis plan and receive prior approval from the scientific steering committee (SQR, LG, JAU, and CT).
The analyses conducted annually include a cross-sectional description of each SMA-type subpopulation. Each subpopulation was delineated in terms of the demographic and clinical characteristics of patients, such as age (current, at diagnosis, at treatment onset, if applicable), sex, familial context, motor, respiratory, and cardiac functions, nutritional and/or ventilatory support, growth scoliosis and orthopedic treatments, and other recognized indicators, alongside DMT use and vital status. Continuous variables were detailed using means and standard deviations, medians, and quartiles, whereas dichotomous variables were presented as proportions. Currently, 4 years post-registry launch, the goal of recruiting over 1000 patients has been met, and we provide a comprehensive cross-sectional description of the enrolled population segmented by SMA type as well as an exploratory survival description using data from both treated and untreated SMA type 1 and 2 subpopulations (crude analysis). For survival description specifically, a patient was deemed treated if she/he had received a) one intravenous dose of OA, b) at least 6 months of daily risdiplam treatment, or c) at least 6 months of nusinersen treatment, inclusive of the initial four loading doses. Kaplan–Meier curves were constructed with 95% confidence intervals and truncated if a sample had fewer than 10 patients at risk. 31 The survival description primarily aims to provide a descriptive, real-world overview of treatment exposure and its evolution over time, rather than a causal comparison of treatment effects.
Results
The results presented here provide an initial descriptive overview of the Registre SMA France, focusing on patient characteristics, treatment patterns, and crude survival descriptive trends. Given the broad scope of the registry, this manuscript does not aim to report on all predefined endpoints but rather to illustrate the types of analyses that can be conducted within this research framework.
Current population overview
As of July 22, 2024, all 64 centers of the French Neuromuscular Network (FILNEMUS) agreed to participate, and 59 centers had already recruited patients. Most centers were pediatric (30) or adult-centered (25), while three covered all ages and one is an intensive care center (htpp://filnemus.fr). Signed informed consent was obtained from 1299 patients for the collection of genetic, clinical, and complementary data. A small number (17, 1.3%) of patients opted for a minimal dataset collection, and six (0.5%) refused to participate in the registry (Figure 1).
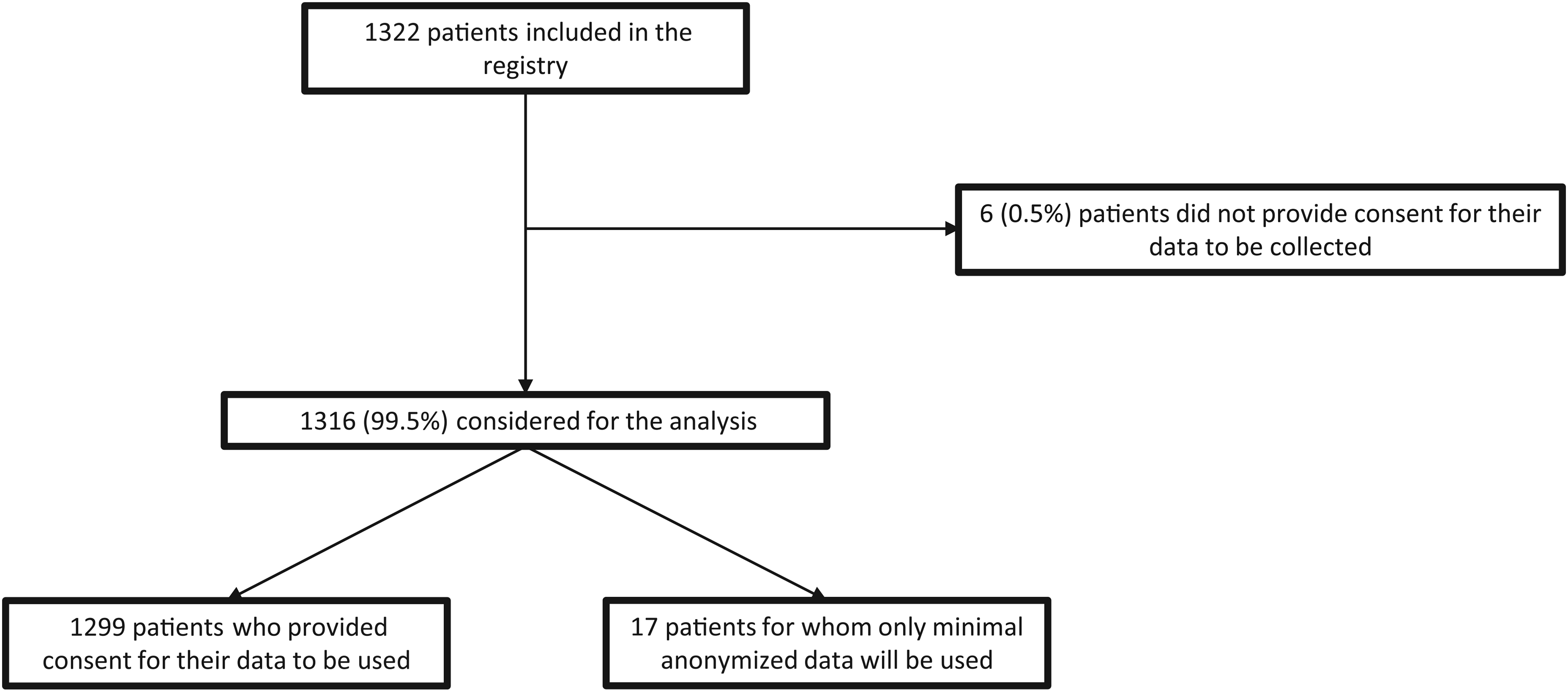
Patient selection.
The COVID-19 pandemic affected enrollment at the launch in 2020, particularly delaying the opening of new participant centers. Therefore, most patients were initially recruited from hospitals in the Parisian region (AP-HP). Four years later (i.e., by 2024), the enrollment rate had stabilized at 15–20 new patients per month nationwide. Figure 2 presents the distribution of treated and untreated patients by SMA type, vital status, and treatment patterns based on registry data extracted for this analysis (July 22, 2024). It is important to note that the registry applies no specific eligibility criteria related to clinical status (e.g., presence of tracheostomy or gastrostomy tube feeding) or prior treatments. Patients were enrolled consecutively, reflecting real-life clinical practices across all participating centers. Among the 1299 analyzed patients, 10 (0.8%) were presymptomatic (SMAp), of whom five were diagnosed presymptomatically due to a family history of the disease, and the remaining five were identified in one of the two regions where an SMA neonatal screening program has been in place since 2023. 26 Family members of > 16% of the patients in the registry were affected by the disease.
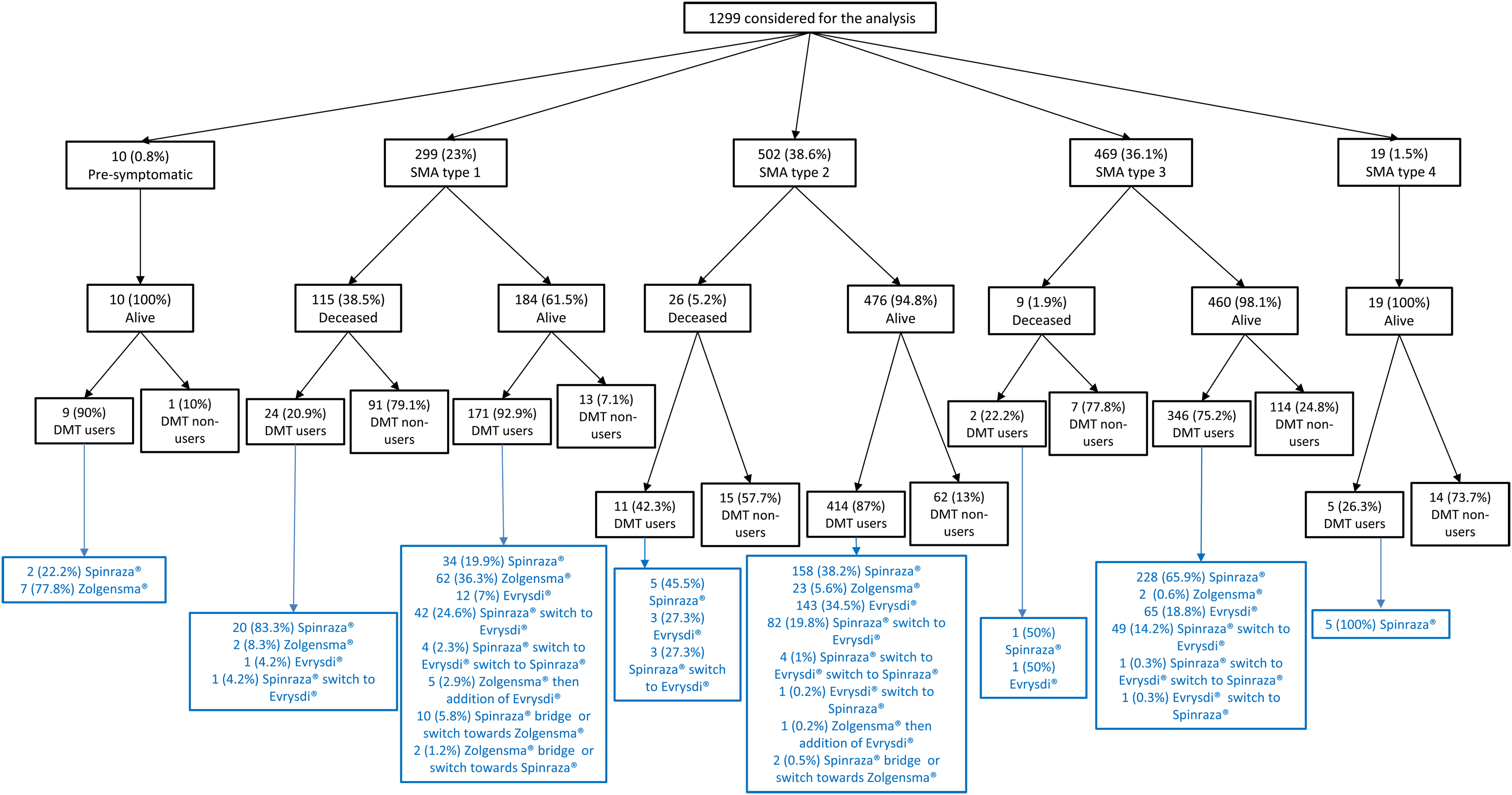
Distribution of patients with SMA according to SMA type, vital status, and DMT use.
Among the symptomatic population, 299 (23%), 502 (38.6%), 469 (36.1%), and 19 (1.5%) had SMA1, SMA2, SMA3, and SMA4, respectively (Figure 2). The demographic, clinical, and diagnostic characteristics of 1270 patients were available. No imbalance in sex distribution was observed across SMA types. Most patients exhibited a homozygous deletion of SMN1, with only 43 carrying a heterozygous deletion and a pathogenic variant (3.4%). The information regarding the number of SMN2 gene copies was available for 1101 patients (86.7%), with most carrying three copies (654 individuals), and the remaining carrying two (232) or four SMN2 copies (199). Only 13 and three patients carried one and five SMN2 copies, respectively (Table 1).
Distribution of patients' characteristics according to SMA type.
Table 1A. Demographic and genetic characteristics.
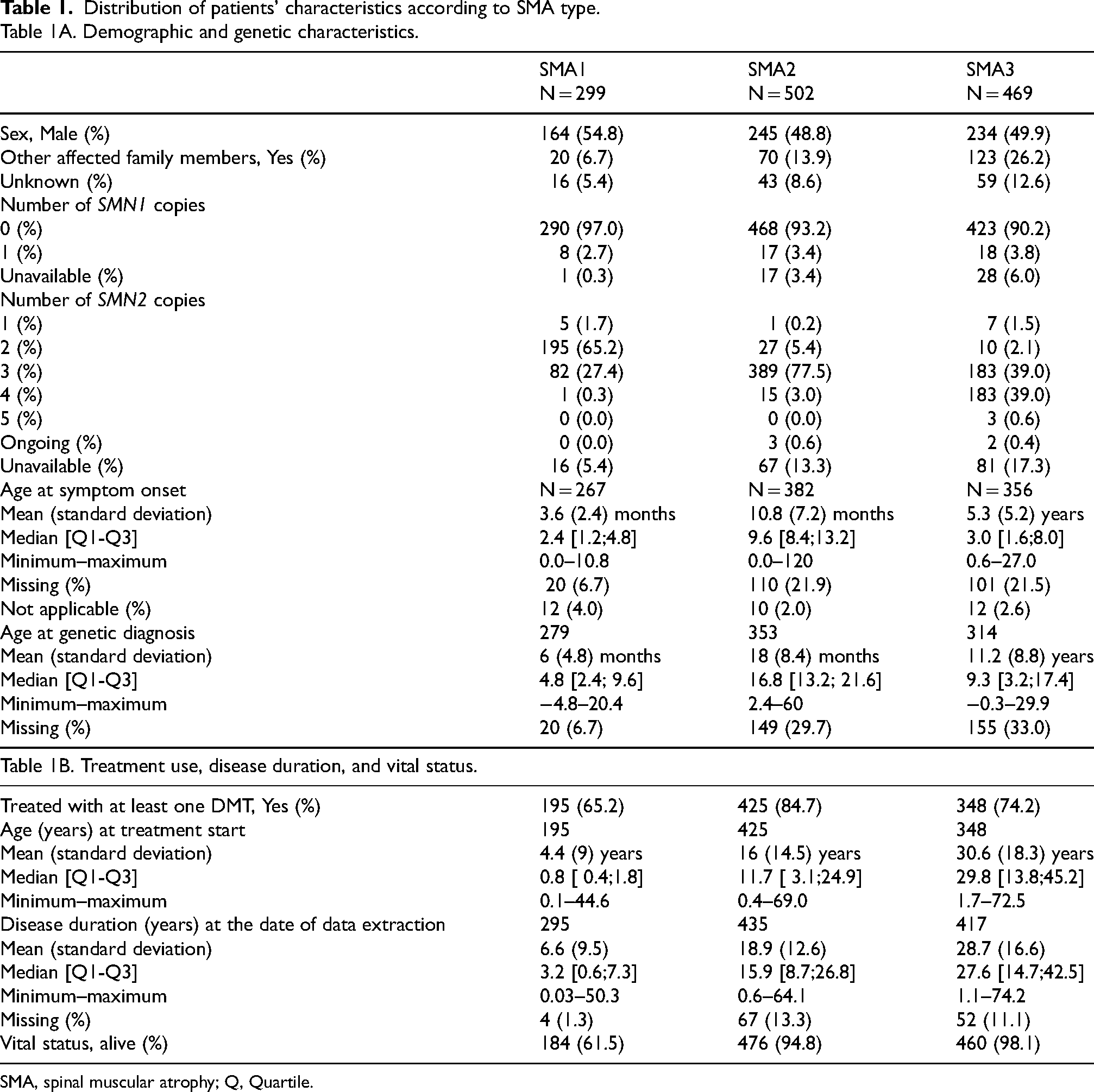
SMA, spinal muscular atrophy; Q, Quartile.
DMT use by SMA type is detailed in Table 1. A total of 968 (76.2%) patients with SMA1, SMA2, and SMA3 received at least one DMT, with treatment rates of 65.2%, 84.7%, and 74.2%, respectively. Most patients who received DMT were treated exclusively with nusinersen (46.1%), whereas 23.2% were treated with risdiplam and 9.2% with OA. Among the 968 treated patients, a total of 208 (21.5%) received more than one DMT, reflecting variable sequences or combinations of therapies. Sequential treatment was the most frequent pattern, observed in 188 patients (19.4%). In most patients (177), the sequence consisted of a switch from nusinersen to oral risdiplam several months or years after the initial intrathecal treatment. Nine patients underwent multiple switches, returning to nusinersen after a period on risdiplam, and two patients switched from risdiplam to nusinersen. In addition, 12 patients (1.2%) received bridging therapy, having initiated treatment with four or fewer doses of nusinersen before transitioning to OA in the following weeks. Combination therapy, defined as the addition of a second DMT to a previously administered one, was observed in 8 patients (0.8%), including 6 who received risdiplam after single-dose OA, and 2 who received nusinersen following OA. . Nine SMAp patients received one DMT (seven, OA; two, nusinersen), and five patients with SMA4 received nusinersen.
The distribution of major disease complications, stratified by SMA type and treatment status, as recorded in the registry on July 22, 2024, is detailed in Table 2. A total of 150 patients (11.5%) had a recorded status of deceased at that point. The vast majority were patients with SMA1 (76.7%) and usually untreated (79%). Only 26 patients had SMA2, and nine had SMA3. Treatment-related death was confirmed in only one case, an infant with SMA1 who died due to thrombotic microangiopathy after treatment with OA.
Distribution of major disease complications in patients according to SMA type, regardless of treatment status.
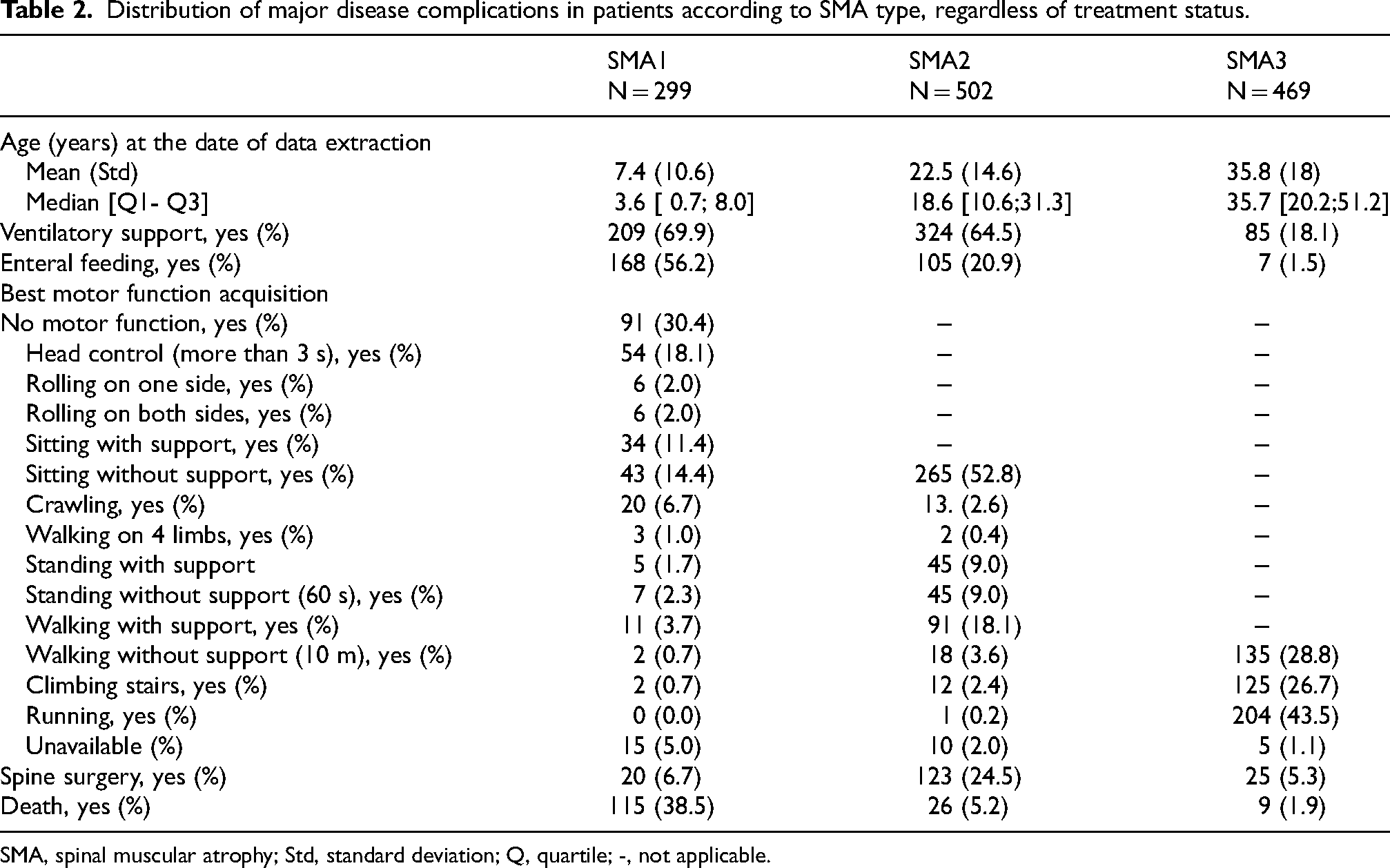
SMA, spinal muscular atrophy; Std, standard deviation; Q, quartile; -, not applicable.
Figure 3 shows the survival rate curves on July 22, 2024, comparing treated vs. non-treated patients with SMA1 and SMA2 (all DMTs considered together), where a drastic change was observed for both treated populations, particularly for infants with SMA1.
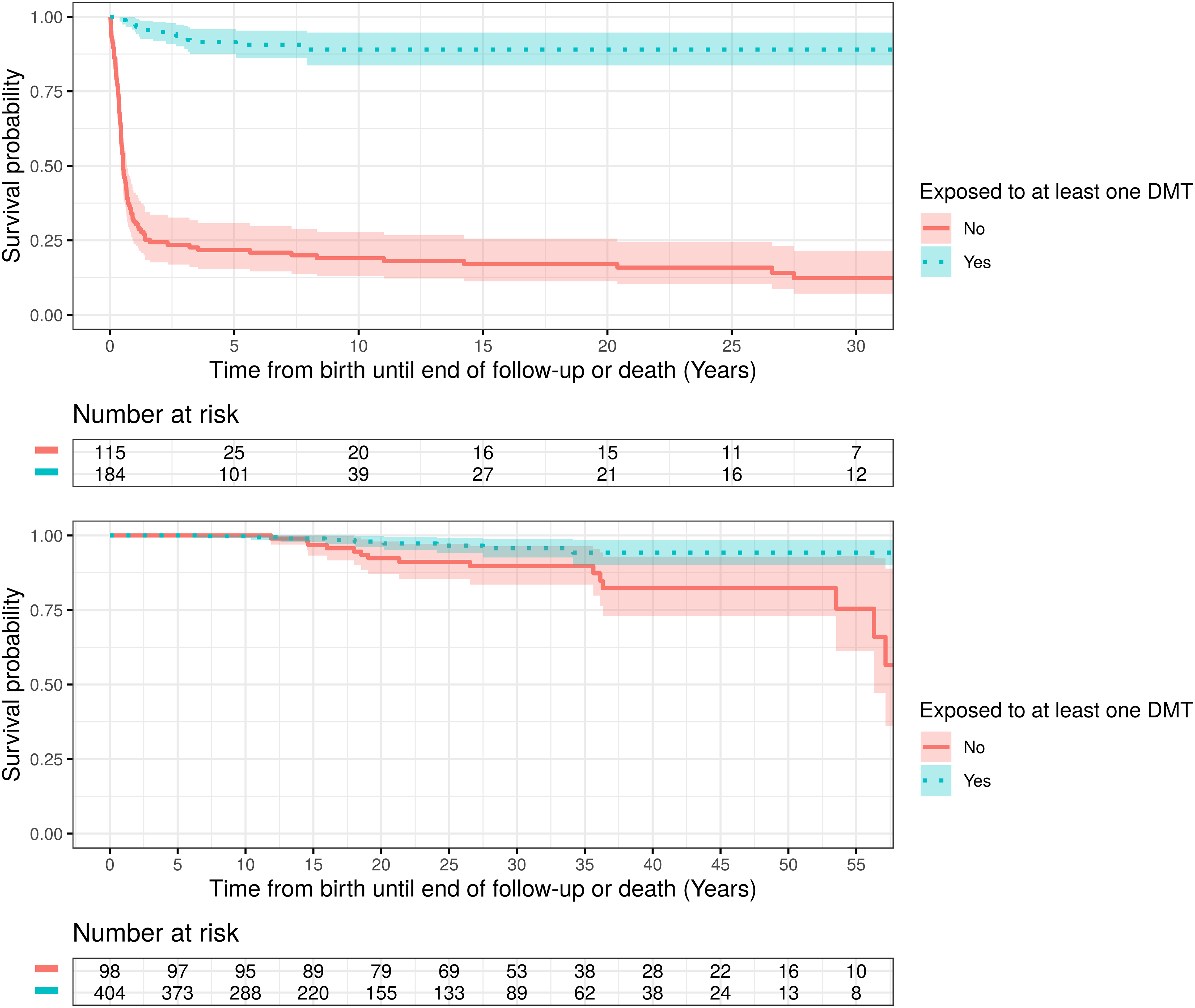
Survival rate curves (Kaplan–Meier curves).
Discussion
The Registre SMA France was established as a comprehensive and adaptable platform for collecting long-term, real-world data on SMA in France. Unlike traditional interventional studies, which focus on predefined patient cohorts and treatment arms, this registry aims to capture the full spectrum of SMA, regardless of disease severity, treatment status, or disease progression. By integrating multicenter and multidisciplinary data, it provides a robust infrastructure to support various research objectives, including natural history studies, post-marketing surveillance of DMTs, comparative effectiveness research, and health policy evaluations.
While the present manuscript provides a descriptive overview of the registry and its current population, it is important to emphasize that its primary purpose is to facilitate ongoing and future research. The dataset continues to expand, with additional modules being developed to address emerging clinical and scientific questions. The value of this registry, therefore, extends beyond the snapshot presented here, as it lays the foundation for a wide range of future analyses that will further advance our understanding of SMA in real-world settings.
As of July 22, 2024, the registry had enrolled 1299 patients across 59 centers, representing 96% of French neuromuscular reference centers. The registry encompasses a highly diverse population, including both treated and untreated individuals, which is essential for evaluating long-term treatment effects and disease evolution in different patient subgroups. The high proportion of patients receiving DMTs (76.2%) highlights the rapid integration of innovative therapies into routine clinical practice in France. Moreover, the registry captures treatment heterogeneity, with 21.5% of treated patients undergoing sequential or combination therapy, reflecting evolving therapeutic strategies and real-world decision-making. Our findings are consistent with data from other international SMA registries, such as SMArtCARE (Germany, Austria, Switzerland), the ISMAC registry (Italy), the RESTORE registry (multinational), and the Canadian Neuromuscular Disease Registry (CNDR).32–36 A Kaplan–Meier survival analysis within our registry shows a significant survival advantage for treated patients, particularly in SMA1 and SMA2, aligning with pivotal clinical trials and previous publications.19,20,37–41 While this analysis was not designed to assess treatment effectiveness, it still highlights a clear survival trend in real-world conditions. However, notable differences exist across registries, particularly regarding treatment strategies. While some registries report the predominance of a single therapy (e.g., nusinersen in SMArtCARE), the French registry shows a more heterogeneous approach, with patients transitioning between therapies or receiving multimodal treatments. This diversity highlights the importance of real-world data in understanding treatment trajectories and optimizing therapeutic strategies.
Although survival has significantly improved with the advent of DMTs, our registry provides critical insights into the evolving natural history of SMA in the treatment era. With longer survival, new clinical challenges emerge, including multisystemic complications and neurocognitive development concerns. Recent reports suggest that some SMA1 survivors experience neurodevelopmental delays, particularly in areas of language and social communication.42,43 To address these concerns, we have developed a dedicated neurocognitive module within our registry to systematically assess these aspects in early-treated children. On another note, the unexpected occurrence of thrombotic microangiopathy following gene therapy prompted the rapid implementation of a biological monitoring module in 2021. 44 This allowed for the identification of early hematological abnormalities, leading to modifications in steroid dosing and monitoring protocols. Since these adjustments, no further deaths related to this complication have been observed, highlighting the critical role of real-world evidence in optimizing patient safety.
The primary strength of the Registre SMA France lies in its comprehensive, historical, and prospective nature, allowing for longitudinal patient follow-up over at least ten years. Its national coverage (96% of centers) ensures that the data collected are highly representative of the real-life SMA population in France. Unlike clinical trials, which are inherently selective, the registry includes all patients with SMA, providing an unbiased and exhaustive perspective on the disease and its management. Furthermore, the registry's flexible structure allows for the integration of new research questions, making it a powerful tool for hypothesis generation. Beyond descriptive analyses, its dataset facilitates comparative effectiveness research, such as assessing the impact of different treatment sequences (monotherapy vs. sequential therapies) and identifying factors influencing treatment response variability. The ongoing expansion of the registry will also provide valuable data to assess the impact of nationwide newborn screening once implemented in France.
Despite its strengths, the registry also faces certain limitations. One challenge is the potential underrepresentation of undiagnosed or less severe cases, as the registry primarily includes patients treated in specialized centers. Additionally, variability in data completeness remains a concern, particularly for untreated patients, where follow-up intervals may be less regular. The heterogeneity in clinical management between centers also introduces variability in treatment and monitoring strategies, which must be accounted for in future analyses.
The Registre SMA France is not only a national database but also an integrated research platform that will support future studies at both national and international levels. A key objective is to compare different treatment strategies, including monotherapy vs. sequential or combination approaches, to determine the most effective long-term therapeutic strategies.
Another major focus will be the long-term safety and durability of DMTs, particularly as more patients transition between therapies or receive treatment from infancy. The registry will also play a key role in evaluating the real-world impact of newborn screening by tracking the evolution of SMA incidence, phenotypic distribution, and long-term outcomes in a post-screening era.
International collaborations with other SMA registries and research networks will be essential for harmonizing data collection and expanding sample sizes for large-scale comparative studies. By integrating our findings with those from other countries, we aim to refine best practices in SMA management and inform public health policies.
Conclusion
This manuscript introduces the Registre SMA France as a dynamic and evolving research tool. The registry provides a comprehensive framework for long-term SMA research, offering real-world insights into disease epidemiology, treatment patterns, and patient outcomes. Its adaptability and extensive national coverage make it a vital resource for future studies exploring treatment effectiveness, safety, and clinical outcomes in SMA. As the registry continues to expand, it will generate valuable data to optimize patient care, support regulatory decisions, and advance scientific knowledge on SMA, both in France and internationally.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602251353446 - Supplemental material for REGISTRE SMA FRANCE: A nationwide observational registry of patients with spinal muscular atrophy in France
Supplemental material, sj-docx-1-jnd-10.1177_22143602251353446 for REGISTRE SMA FRANCE: A nationwide observational registry of patients with spinal muscular atrophy in France by Lamiae Grimaldi, Rocio Garcia-Uzquiano, Marta Gomez-Garcia de la Banda, Amal Oulhissane-Omar, Celine Tard, Pascale Saugier-Veber, Vincent Laugel, Isabelle Desguerre, Pascal Cintas, Carole Vuillerot, Frederic Audic, Claude Cances, Tanya Stojkovic, Jon Andoni Urtizberea, Shahram Attarian, Juliette Ropars, Susana Quijano-Roy and the Registre SMA France Study Group in Journal of Neuromuscular Diseases
Supplemental Material
sj-xlsx-2-jnd-10.1177_22143602251353446 - Supplemental material for REGISTRE SMA FRANCE: A nationwide observational registry of patients with spinal muscular atrophy in France
Supplemental material, sj-xlsx-2-jnd-10.1177_22143602251353446 for REGISTRE SMA FRANCE: A nationwide observational registry of patients with spinal muscular atrophy in France by Lamiae Grimaldi, Rocio Garcia-Uzquiano, Marta Gomez-Garcia de la Banda, Amal Oulhissane-Omar, Celine Tard, Pascale Saugier-Veber, Vincent Laugel, Isabelle Desguerre, Pascal Cintas, Carole Vuillerot, Frederic Audic, Claude Cances, Tanya Stojkovic, Jon Andoni Urtizberea, Shahram Attarian, Juliette Ropars, Susana Quijano-Roy and the Registre SMA France Study Group in Journal of Neuromuscular Diseases
Footnotes
Acknowledgments
The authors gratefully acknowledge the patients for their participation and the pharmaceutical companies (Biogen, Novartis Gene Therapies, and Roche) for their financial support. We also appreciate the team of the Paris-Saclay Clinical Research Unit at the Assistance Publique-Hôpitaux de Paris (AP-HP), particularly Mme Lionelle Nkam, Senior Statistician, Mme Bertille Alauze, Project Manager of the Registry, and Mme Nawal Derridj, Senior Project Manager, for their expertise and involvement in both the registry and manuscript preparation. We acknowledge all the technical research assistants who worked at the participating sites, as well as the clinicians and caregivers involved in the care of French SMA patients at the reference centers. We also thank the French Neuromuscular Network (FILNEMUS) and the European ERN Network Euro-NMD.
ORCID iDs
Ethical considerations
The registry protocol was approved by the Institutional Ethical Review Board of the Assistance Publique-Hôpitaux de Paris, which was also responsible for data processing (protocol number: APHO200085).
Consent to participate
Participants (either parents or legal caregivers, in the case of minors) provided signed informed consent for their health data to be used in this study.
Author contributions
All authors contributed to the writing or reviewing of the protocol, selection of variables, scales, and patient outcomes for inclusion in the registry. All authors also reviewed the manuscript. L.G., R.G.U., and S.Q.R. wrote the manuscript, analyzed the data presented, and conducted a thorough review.
Funding
This registry is sponsored by Assistance Publique-Hôpitaux de Paris and supported by unrestricted grants from Biogen, Roche, and Novartis.
Declaration of conflicting interests
S.Q.R, M.G.G.B, V.L, I.D, C.C, S.A, and J.R have served on advisory boards and received consulting fees, travel reimbursements, and institutional grants from Avexis, Biogen, Novartis, and Roche. I.D, J.A.U, and J.R has served on the advisory board for Pfizer and I.D has received travel reimbursements and consulting fees. R.G.U. has served on advisory boards and received consulting fees and travel reimbursements, from Roche. C.T has received consultant fees and non-financial support from Sanofi-Genzyme, Akcea, Pfizer, Alnylam, Ultragenyx, Biogen, Santhera, LFB, Argenx, UCB, Amgen, Lupin, Novartis, Roche, Astra-Zeneca and CSL Behring. S.A has received consultant fees and non-financial support from Johnson & Johnson, Pfizer, Alnylam, Biogen, LFB, Argenx, UCB, Amicus, Novartis, Roche, Sanofi, Alexion and Astra-Zeneca. P.S.V has received consultant fees from Novartis and grants from Roche, ABM and Filnemus, and travel reimbursement from ENM. P.C, T.S has received consulting fees from Biogen and Roche. C.V and J.A.U have served on advisory boards and received consulting fees, travel reimbursements, and institutional grants from Novartis and Roche. F.A has served on advisory boards and received consulting fees, travel reimbursements, and institutional grants from Novartis and Biogen. J.A.U has received consultant fees from AFM-Telethon. L.G and A.O.O declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.