
Abstract
Genetically engineered animal models are major tools of a drug discovery pipeline because they facilitate understanding of the molecular and biochemical basis of disease. These highly complex models of human disease often require increasingly convoluted genetic analysis. With growing needs for throughput and consistency, we find that traditional aspiration-and-dispense liquid-handling robots no longer have the required speed, quality, or reproducibility.
We present an adaptation and installation of an acoustic droplet ejection (ADE) liquid-handling system for ultra-high-throughput screening of genetically engineered models. An ADE system is fully integrated with existing laboratory processes and platforms to facilitate execution of PCR and quantitative PCR (qPCR) reactions. Such a configuration permits interrogation of highly complex genetic models in a variety of backgrounds. Our findings demonstrate that a single ADE system replaces 8–10 traditional liquid-handling robots while increasing quality and reproducibility.
We demonstrate significant improvements achieved by transitioning to an ADE device: extremely low detectable cross-contamination in PCR and qPCR despite extensive use, greatly increased data reproducibility (large increases in data quality and Cq consistency), lowered reaction volumes for large cost savings, and nearly a magnitude increase in speed per instrument. We show several comparisons between traditional- and ADE-based pipetting for a qPCR-based workflow.
Introduction
The use of laboratory animals in research drives functional genomics and the community’s understanding of gene function through organismal mutagenesis. 1 By manipulating animal genomes, researchers can elucidate the etiology of cancer and other highly impactful human diseases. 2 Mice, in these settings, are often used as a model organism because there is translational correlation between observations recorded from investigation of a mutant mouse to the potentiality of producing a marketable human drug treatment based on such data. 3 Moreover, the mouse is a practical research animal: It is one of the smallest mammals, has a short generation time of 10 weeks from being born to giving birth, and is typically a prolific breeder generating 5–10 pups per litter. 4 Scientists are routinely able to replace endogenous mouse genes for their human counterparts, thus “humanizing” them, allowing researchers to generate results more directly applicable to human disease.5,6
Changes to a mouse’s genome can be introduced through the manipulation of embryonic stem cells to create chimeric mice, some of which will possess the ability to potentiate the introduced mutation through germline breeding. 7 These changes have been classically introduced by random chemical mutagenesis or through pronuclear injection of a foreign transgene that is randomly integrated into the genome. 8 For the past 25 years, locus-specific genomic manipulation has been carried out by electroporating mouse embryonic stem cells with a DNA construct containing flanking homology arms that drive incorporation into the genome via homologous recombination pathways. 9 Use of the Cre/loxP or Tet-On/Tet-Off systems enables creation of conditionally expressive models, allowing researchers to control both spatial and temporal expression of modified loci.10–12 Novel and emerging techniques use nucleases conjugated to a specific DNA binding domain, such as zinc-finger nucleases 13 or the Cas9/CRISPR system. 14 Of particular interest, CRISPR has been proven to create multiple mutations in a mouse in a single step 15 and has been demonstrated to create a variety of construct types. 16
Modified organisms require careful and accurate genotyping to identify founding animals and continue production through a colony’s lifetime. Like any large-scale effort with high degrees of complexity, such operations directly benefit from laboratory automation efforts. Lab robotics have been used previously to carry out PCR-based genotyping of mice, 17 SNP typing of blood 18 and forensic 19 samples, high-resolution melting experiments, 20 characterizing mutated yeast strains, 21 genotyping bacterial pathogens, 22 performing DNA extraction, 20 and automating sequencing of human disease loci. 23 We use lab automation systems to carry out the bulk of our laboratory’s work: the extraction of gDNA from ~350,000 samples to assemble >1 M PCR and qPCR reactions to facilitate analysis of ~800,000 genotypes per annum.
Previously, we have relied on traditional liquid-handling robots, using standard aspirate-and-dispense pipetting technology, to assemble our molecular amplification reactions. We found that increasing workloads do not scale well on these platforms; more work requires the installation of more robots, consuming a good deal of resources (i.e., lab space and funding). In previous observations and detailed here, we find these robots do not always produce the precision desired, nor do they rigorously prevent cross-contamination between samples. Moreover, there has been little transformative progress in aspirate-and-dispense technology. Although incremental progress continues to be made, the landscape of using robots to transfer liquids using disposable or fixed tips has changed little in the past 15 years. The introduction of acoustic droplet ejection (ADE) pipettors drastically changes this landscape.
The history and physics of ADE are well documented in this journal issue. In brief, energy is acoustically transferred from a transducer into a microplate containing reagent to be transferred. This focused energy results in the ejection of a small droplet (here, 25 nl) that travels directly upward into an upside-down recipient plate, which captures the droplet via surface tension. 24 Hundreds of droplets can be fired each second and flown in a very precise manner without any physical contact being made between the device and the reagent, greatly reducing cross-contamination. 25 Recent developments to ADE liquid handlers in the past two years have made these devices accessible to the genomic arena.
We have installed and used ADE lab robotics to carry out the genotyping of several hundred thousand genetically engineered rodents. Genotyping is carried out across two platforms: PCR-based fragment analysis and real-time or quantitative PCR (qPCR). Below, we present our initial testing and evaluate this liquid handling platform in the context of increasing precision while simultaneously reducing cross-contamination to a great extent. In addition, we discuss other benefits by adopting this technology: very large throughput increases, reduction of primer and probe handling, and potential applications of this technology for other molecular biology–based applications.
Materials and Methods
Robotic Assembly Platforms
Aspirate-and-dispense liquid-handling robot: Tecan Evo robots were used as representative traditional laboratory pipetting robots and were outfitted with standard diameter system tubing, 0.5 ml syringes, and Span-8 Teflon-coated fixed tips. Tips are cleaned between reagents via dispensing of 50 ml distilled water into the wash station, followed by tip washing with 5 ml distilled water into the shallow wash station cleaner. System tubing is scrubbed with detergent, and fixed tips are bleach-treated according to the manufacturer’s instructions. The Evo worksurface is outfitted with labware to hold Eppendorf tubes containing master mix and assays, gDNA source plates, and 96-well destination PCR plates.
Acoustic droplet ejection pipettor and supplementary robotics: Labcyte Echo 525s were chosen to assemble qPCR reactions by using acoustic droplet ejection. Our configuration contains two Echo 525s accessed by an Access robotic arm workstation ( Fig. 1 ). The Access deck houses peripherals necessary for an unattended amplification reaction assembly: a Thermo-Fisher Scientific Combi NL Universal for bulk dispensing of mastermix and water, a Nexus Xpeel microplate desealer to remove microplate adhesive seals, an Agilent PlateLoc Sealer to seal fully assembled PCR plates, and an Agilent Microplate Centrifuge to reset reagent menisci before acoustic transfer.
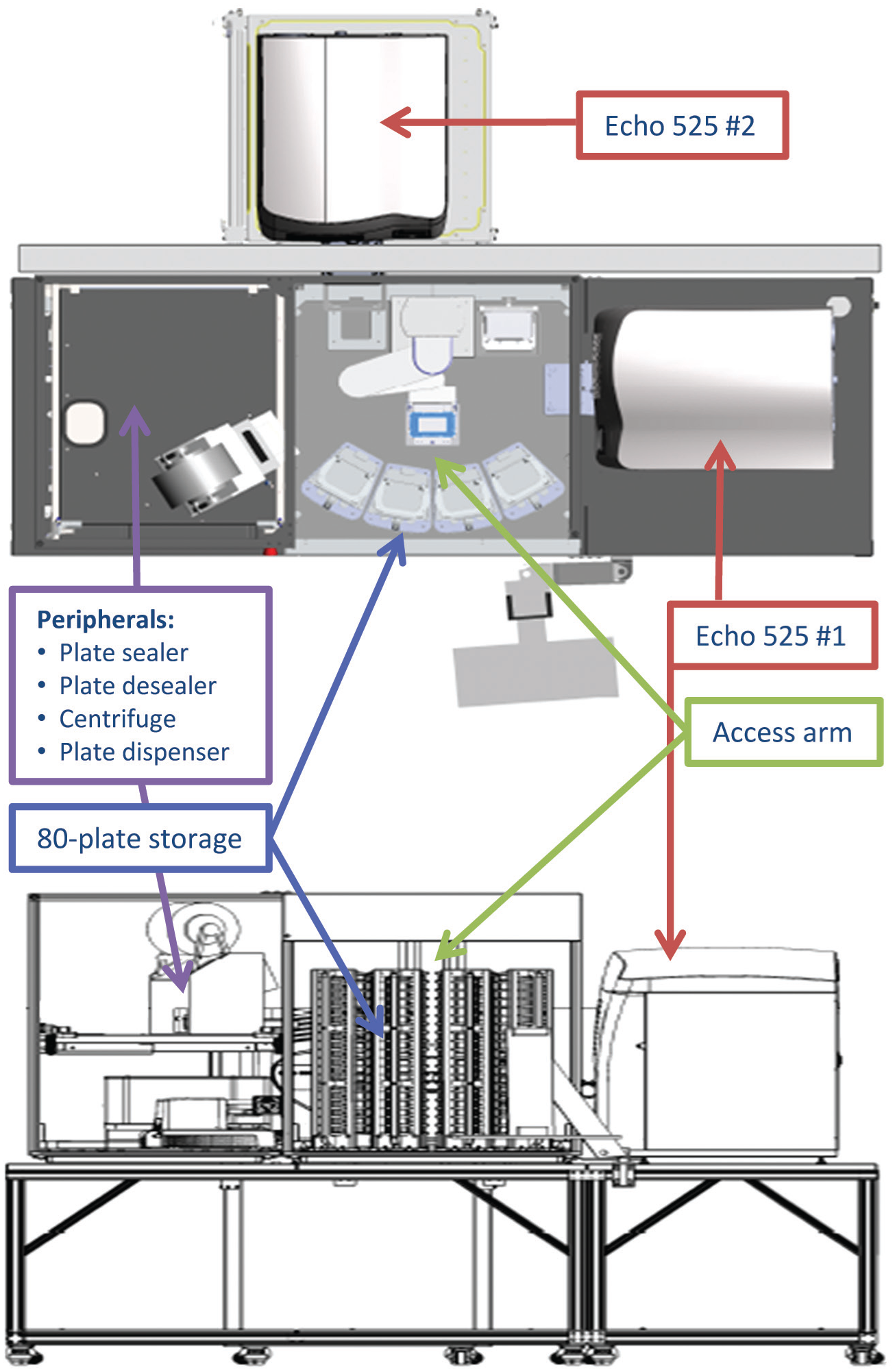
Schematic of a high-throughput mammalian genotyping reaction assembly platform using acoustic droplet ejection. Shown is a top-down view (top) and a front-facing view (bottom). The platform consists of two Labcyte Echo 525s serviced by an Access robotic arm. In addition to providing reagent delivery to the acoustic dispensers, the arm also serves accessory peripherals: a microplate sealer, microplate desealer, microplate centrifuge, low-volume bulk plate dispenser, and microplate storage hotels.
Precision Testing with a qPCR Platform (Rosa26 Locus)
A pool of gDNA was created from extractions of ~1000 wild-type mice from preps created using the DNeasy 96 Blood & Tissue Kit (Qiagen, Venlo, the Netherlands). gDNA template concentrations in these reactions on the Evo (20 µl) and Echo (5 µl) platforms were approximately 8 µM. Reactions were assembled in the following manner: 400 nM forward and reverse primers, 150 nM probe, and 1× Type-It Fast SNP PCR Master Mix + ROX dye (Qiagen). Nucleotide sequences for these oligos are as follows: Rosa26WT forward: CCCGCCC ATCTTCTAGAAAGA; Rosa26WT reverse: CTGGGCCTG GGAGAATCC; and Rosa26WT probe: /56-FAM/TTCCCCCTCTTCCCTCGTGATCTGC/3IABkFQ/. The endogenous control assay is composed of the following: ApoB forward primer: CACGTGGGCTCCAGCATT; ApoB reverse primer: TCACCAGTCATTTCTGCCTTTG (oIMR1544, oIMR3580; Jackson Laboratory, Bar Harbor, Maine); and ApoB probe: VIC-CCAATGGTCGGCACTGCTCAA-MGBNFQ (ABI/Life Technologies, Carlsbad, CA). No-template-control (NTC) reactions contained distilled water in place of gDNA template. Assays were cycled on a 7900HT Fast Real-Time PCR System (ABI/Life Technologies) and were analyzed using SDS 2.4.1 (ABI/Life Technologies). Assays were thermal cycled as follows: initial denaturation (95 °C, 5′) followed by 35 cycles of PCR (95 °C, 60″; 60 °C, 30″; and 72 °C, 60″).
Contamination Testing with a qPCR Platform (Cre Locus)
Genomic DNA from Cre+ positive mice were purified using the Agencourt DNAdvance kit (Beckman Coulter, Brea, CA) executed on a Biomek FXP liquid handler (Beckman Coulter). gDNA from 560 animals all harboring a Cre transgene were pooled to make a Cre+ positive gDNA “pooled” stock. In addition, 192 mouse gDNA samples were identified as Cre-harboring samples to represent individual (versus pooled) samples. qPCR assays were assembled on the Tecan Evo in 96-well plates to a final volume of 20 µl; the Labcyte 525 was used to construct reactions in a 384-well plates to a final volume of 5 µl. Experimental and NTC reactions were arranged in an alternating checkerboard pattern. gDNA template concentrations in these reactions on the Evo and Echo platforms were approximately 8 µM and 5 µM, respectively.
The reactions were assembled in the following manner: 400 nM forward and reverse primers, 150 nM probe, and 1× Type-It Fast SNP PCR Master Mix + ROX dye (Qiagen). Nucleotide sequences for these transgene oligos are as follows: Cre forward primer: GCGGTCTGGCAGTAAAAACTATC; Cre reverse primer: GTGAAACAGCATTGCTGTCACTT (oIMR1084, oIMR1085; Jackson Laboratory); and Cre probe: FAM/AA+ACATGC+T/ZEN/TCA+TCG+TCGG/3IABkFQ (a plus sign preceding the nucleotides indicates LNA bases; IDT, Coralville, IA). The endogenous control assay was composed of the following: ApoB forward primer: CACGTGGG CTCCAGCATT; ApoB reverse primer: TCACCAGTCATT TCTGCCTTTG (oIMR1544, oIMR3580; Jackson Labora-tory); and ApoB probe: HEX/CC+A+ATGG+TC/ZEN/GGGCAC+TG/3IABkFQ (IDT; a plus sign preceding the nucleotides indicates LNA bases). NTC reactions contained distilled water in place of gDNA template. Assays were cycled on a C1000 CFX Touch optical thermalcycler (BioRad) and were analyzed using CFX Manager 3.1 (BioRad). Assays were thermal cycled as follows: initial denaturation (95 °C, 5′) followed by 50 cycles of PCR (95 °C, 60″; 60 °C, 30″; and 72 °C, 60″).
Contamination Testing with a PCR / Fragment Analysis Platform (PS2 Locus)
Genomic DNA from PS2+ positive mice were purified using the Agencourt DNAdvance kit (Beckman Coulter) executed on a Biomek FXP liquid handler (Beckman Coulter). gDNA from 143 animals all harboring a PS2 transgene were pooled to make a PS2+ positive gDNA “pooled” stock. qPCR assays were assembled on the Tecan Evo in 96-well plates to a final volume of 20 µl; the Labcyte 525 was used to construct reactions in 384-well plates to a final volume of 5 µl. Experimental and NTC reactions were arranged in an alternating checkerboard pattern. gDNA template concentrations in these reactions on the Evo and Echo platforms were approximately 8 µM and 5 µM, respectively.
The reactions were assembled in the following manner: 625 nM forward and reverse primers and 1× Type-It Fast SNP PCR Master Mix + ROX dye (Qiagen). Nucleotide sequences for these oligos are as follows: PS2 forward primer: /56-FAM/TCATTGGCTTGTGTCTGACCCT; PS2 reverse primer: GCTTTCAACGTCAGTAGGACAA; IL8R forward primer: /56-FAM/CTTCGCTGTCGTCCTTGTCT; and IL8R reverse primer: AGCCATGATCCTGAGAAGTC CAT. NTC reactions contained distilled water in place of the gDNA template. 96-well Evo-assembled assays were cycled on a GeneAmp PCR 9700 System (ABI/LifeTech), whereas Echo-assembled reactions were cycled on a C1000 CFX Touch.
Assays were thermal cycled as follows: initial denaturation (95 °C, 5′) followed by 30 cycles of PCR (95 °C, 60″; 60 °C, 30″; and 72 °C, 60″). PCR assays were analyzed using a 3730XL DNA Analyzer (ABI/Life Technologies). POP-7 Performance Optimized Polymer (ABI/Life Technologies) was used as a matrix within a 50 cm capillary array. GeneScan 600 LIZ size standard (ABI/Life Technologies) was used as a system control; a 30× dilution of the size standard was carried out in HiDi Formamide (ABI/Life Technologies) and used to dilute PCR samples by 21×. Ten microliters of the diluted assays were loaded onto the DNA Analyzer. Data were analyzed using Gene Mapper 4.0 software (ABI/Life Technologies).
Results and Discussion
Evaluation of Data Consistency
A traditional liquid handler was directly compared to an acoustic-based dispenser to evaluate qPCR reaction assembly. Initially, we evaluated the consistency and robustness of analyzing large cohorts of genetically modified mice on each platform. We randomly chose a large cohort of mice containing ~1000 individuals in roughly a dozen colonies containing the same modified locus. After pooling those samples together to create a “master” DNA source, we then set up qPCR reactions containing primer and probes to interrogate two endogenous control genes (Rosa26 and ApoB). Care was taken to minimize sources of variability that could be imparted from other aspects of reaction assembly, such as those attributable to using different lots of assay reagents, assembling master mixes in multiple sessions, and so on. Reactions were assembled as described and cycled 35 times while monitoring accumulation of probe signals.
Results are depicted in Figure 2 ; amplification curves are shown for each probe channel [i.e., VIC fluorophore (Apo) and FAM fluorophore (Rosa26)] plotted for each robotic assembly platform ( Fig. 2a ). These same data are presented as an endpoint scatterplot in which the Rosa26 signal is plotted on the X-axis and the ApoB probe signal on the Y-axis ( Fig. 2b ). Data resulting from ADE-based assembly demonstrate a higher level of precision than that of traditionally assembled reactions in both amplification curves and scatterplots. Specifically, amplification curves ( Fig. 2a ) for the ADE-assembled reactions demonstrate more similar Cq values than those assembled with a classical liquid handler (summarized in Table 1 ). In this experiment, both detectors displayed superior standard deviations between samples when assembled by ADE versus traditional robots. This is one of many illustrations of an increase in precision and reproducibility when applying ADE technology to genomic applications.
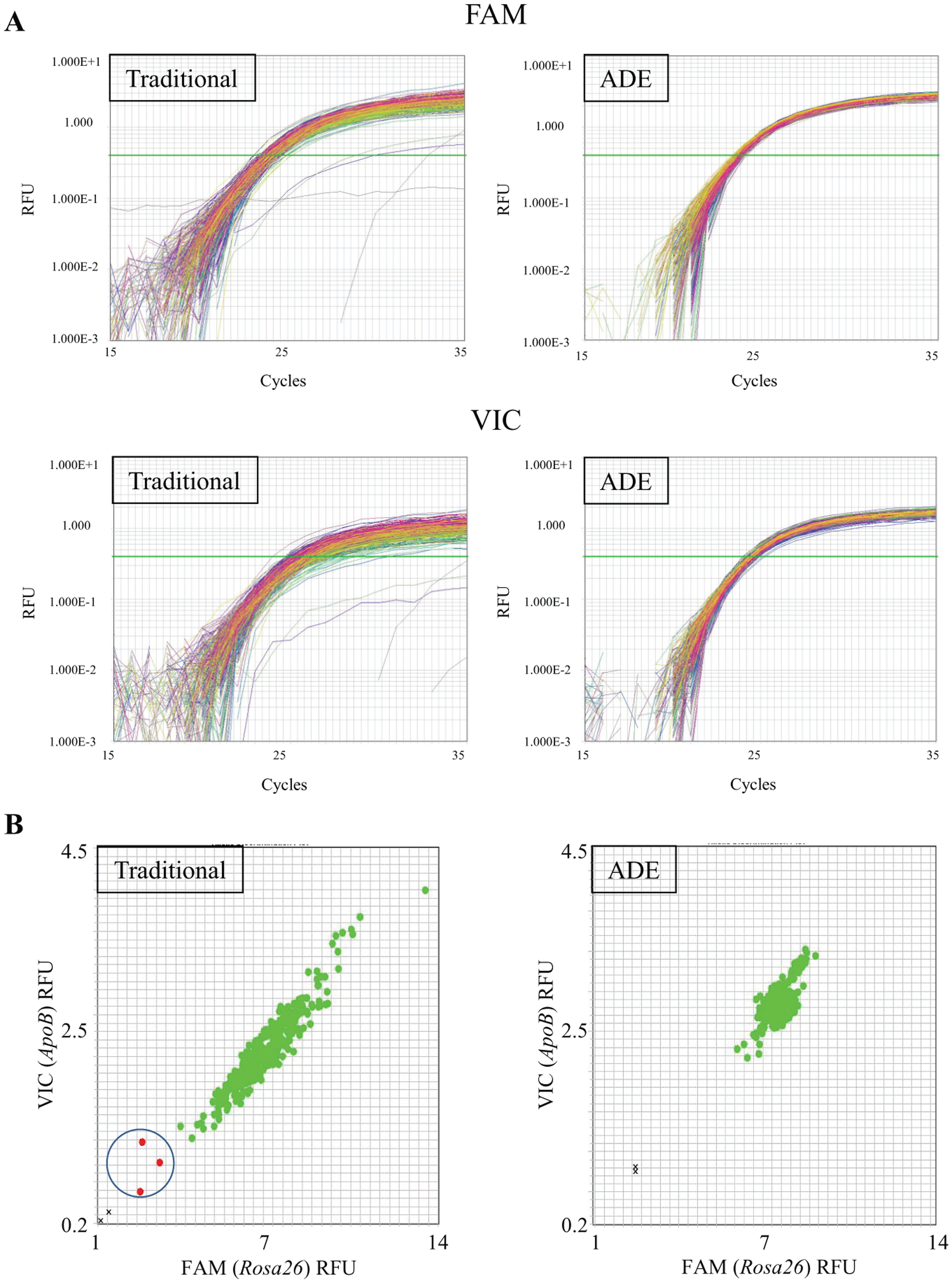
Comparison of quantitative PCR (qPCR)-based genotyping data quality by platform. Amplification curves (A) and cluster plots (B) are depicted for both traditional- and acoustic droplet ejection (ADE)-assembled reactions. (
Summary of qPCR Quality Metrics by Assembly Platform.
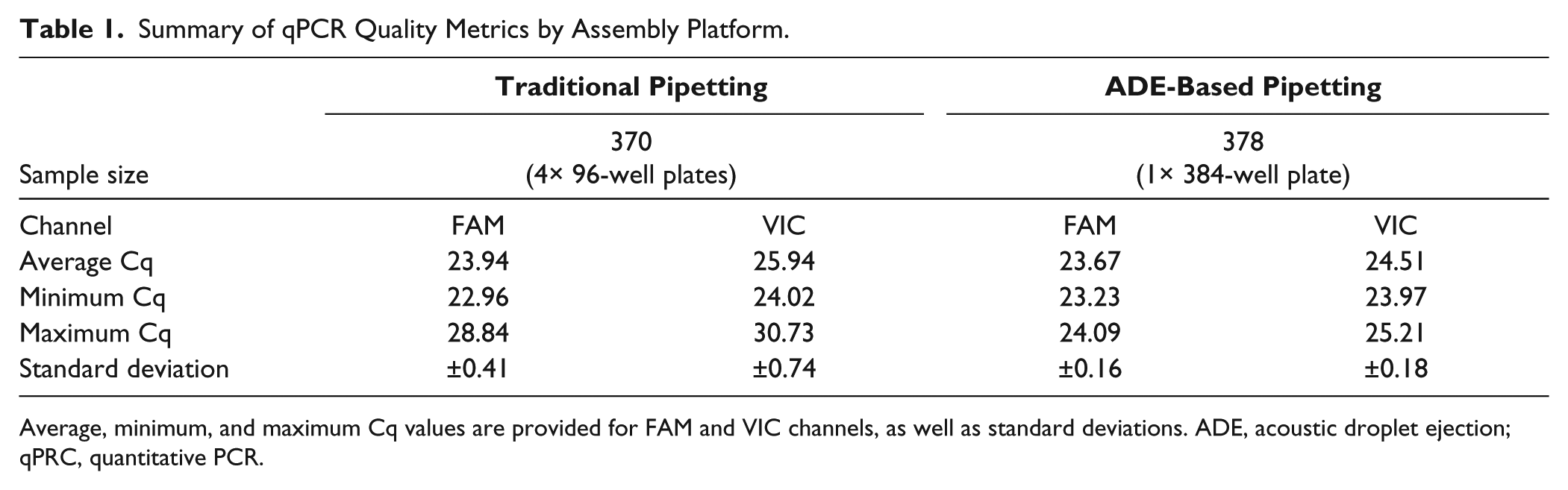
Average, minimum, and maximum Cq values are provided for FAM and VIC channels, as well as standard deviations. ADE, acoustic droplet ejection; qPRC, quantitative PCR.
Differences in data quality are more apparent when viewed as a scatterplot ( Fig. 2b ). Reactions created by traditional pipetting platforms demonstrate a greater variability in the scattercloud (left) than with ADE-assembled reactions (right). The ratiometric distance between the NTC signals (black crosses) and the lowest positive RFU values (green circles) are nearly doubled between the two platforms for both probes. Moreover, three data samples from the classically assembled reactions are ambiguous and require reanalysis; zero samples from the ADE platform were seen to be uncertain. In the context of a mammalian genotyping laboratory, this is highly impactful. Unlike cell-based work in which it is commonly accepted that some data points will drop out due to the nature of high-throughput workflows, this remains an intractable issue in a vivarium setting. Compassionate welfare standards and strict adherence to the 3R principles26–28 and Institutional Animal Care and Use Committee procedures dictate that, in many cases, an animal’s genotype must be evaluated to properly determine the appropriate course of action for that individual. Significant numbers of samples requiring reanalysis not only create additional burden and extended turnaround times for a genetic analysis laboratory but also increase the workload of a colony resource and management team caring for the genetic models. Reduction of samples requiring reanalysis is an understated boon when performing analysis of animal models. Overall, improved clustering yields not only a reduced reanalysis load but also more rapid and straightforward cohort-based data analysis.
Evaluation of Cross-Contamination in Quantitative PCR
One of the many compelling reasons to transition from a classic to ADE-based reaction setup is the giant reduction in observable cross-contamination.29,30 In our first tests with acoustic-based dispense technology (data not shown), we examined cross-contamination in limited fashion at 35 amplification cycles. Our initial results were promising: 16 of 96 (16.7%) NTC reactions assembled by a traditional laboratory robot were found to be contaminated with exogenous gDNA, whereas none of 168 reactions (0%) assembled by ADE demonstrated any apparent cross-contamination. Because these data seemed so remarkable, we wished for a more thorough investigation and so initiated the following study.
We performed this study to evaluate cross-contamination in two different modes. To examine only platform-to-platform variability, we ran several series of reactions composed of a single source of gDNA pooled from a large cohort of mice all sharing the same modified locus. This facilitates simple comparisons between platforms. To better mirror actual mammalian genotyping operations, we repeated these series using individual and unique gDNA samples. This was done to account for the animal-to-animal and cohort-to-cohort variability that is observed when genotyping inbred laboratory strains. We identified many individual gDNA samples containing the same transgene (Cre recombinase) under various endogenous mammalian promoters and probed for it using an endogenous locus (ApoB) as a positive control. The result of this project is shown in
Figures 3
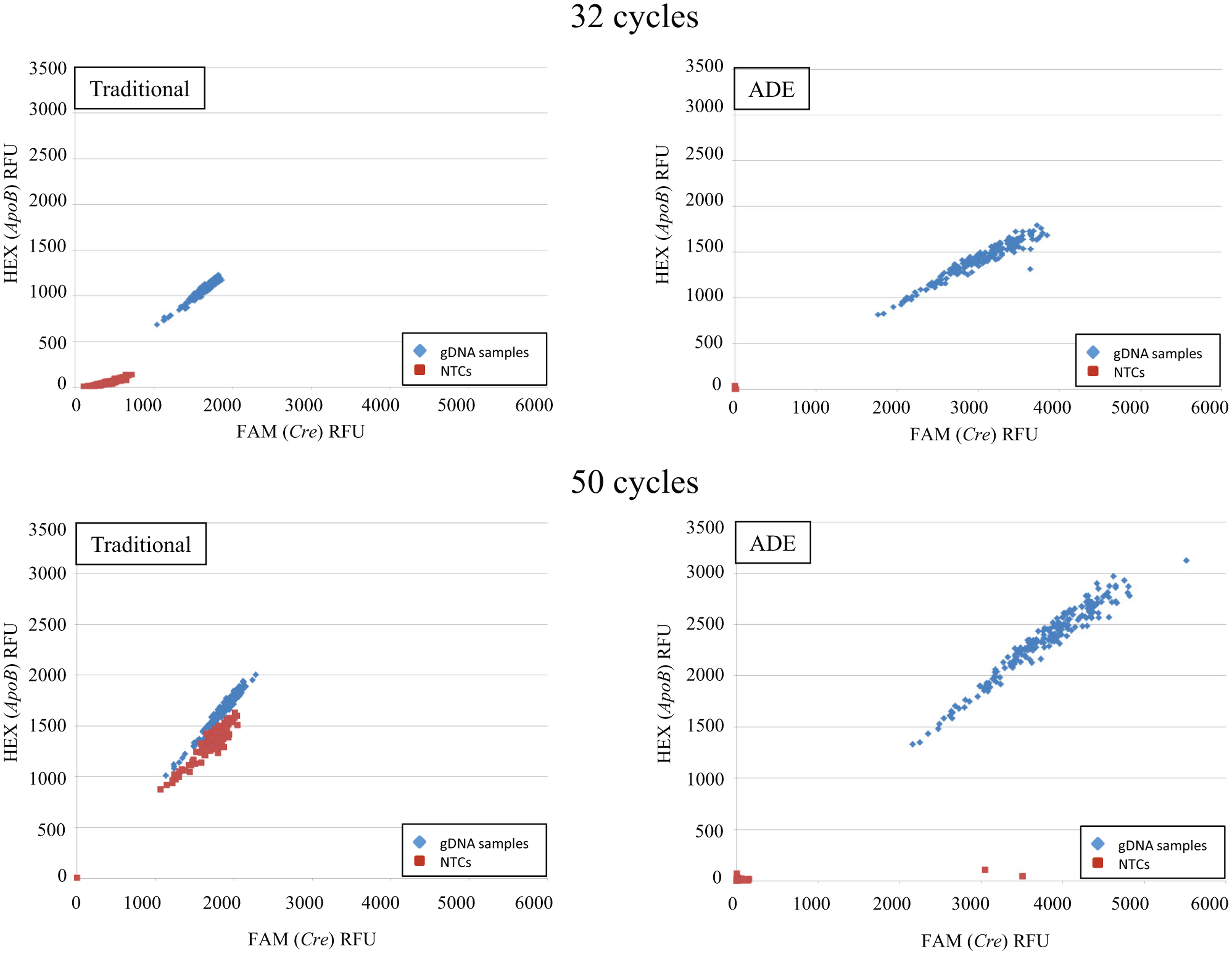
Comparison of contamination observed when performing quantitative PCR (qPCR)-based genotyping using a single, pooled gDNA sample. Cluster plots are shown for 32 cycles of amplification (top) and 50 cycles (bottom). Reactions were assembled by a traditional robot (left column) or an acoustic droplet ejection (ADE) robot (right column). Blue dots represent positive samples; red dots are no-template controls (NTCs).
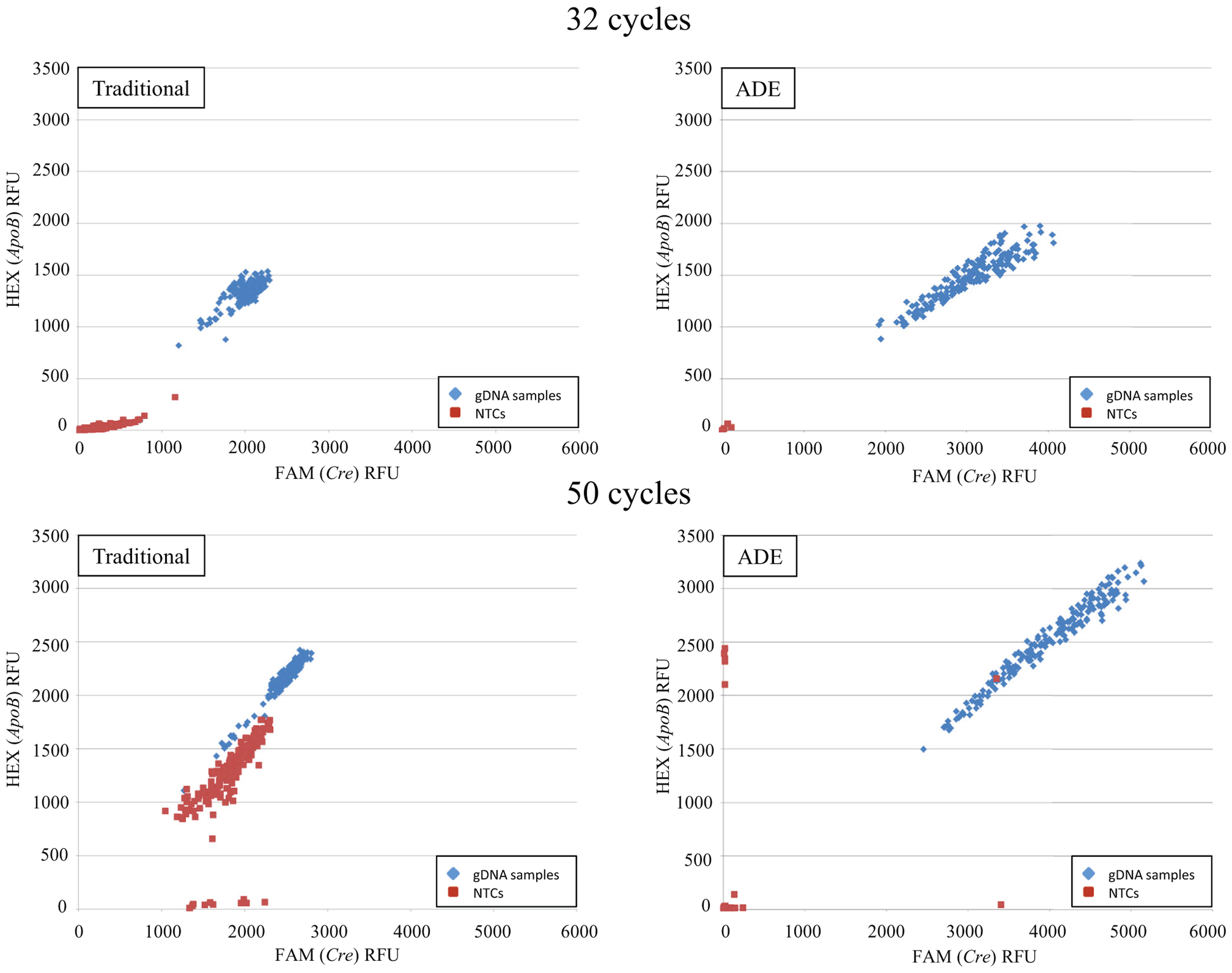
Comparison of contamination observed when performing quantitative PCR (qPCR)-based genotyping using many individual gDNA samples. Cluster plots are shown for 32 cycles of amplification (top) and 50 cycles (bottom). Reactions were assembled by a traditional robot (left column) or an acoustic droplet ejection (ADE) robot (right column). Blue dots represent positive samples; red dots are no-template controls (NTCs).
Scatterplots are displayed at a typical amount of PCR amplification (i.e., 32 cycles; Fig. 3 , top row) and an extreme amount of cycling to clearly observe contamination (50 cycles; Fig. 3 , bottom row). Reactions assembled using classical robotics ( Fig. 3 , left column) are compared to ADE-assembled reactions ( Fig. 3 , right column). The results between platforms are striking. In viewing the comparison between platforms at 32 cycles, the ADE-assembled reactions show no sign of amplification in NTC controls (red dots, N = 192). In contrast, nearly all (97.9%) NTC reactions created by the traditional pipetting platform demonstrated appreciable levels of amplification. At certain levels, this causes reanalysis of certain cohorts when a particular NTC is no longer statistically significant from the positive amplification signals in that grouping. In addition, the relative signal intensity is nearly double with the ADE-created reactions interrogating the noncontrol probe (Cre, X-axis). Once again, we observe a large and meaningful increase in the signal-to-noise (S:N) ratios between the different pipetting technologies.
This trend is more evident at very high levels of amplification (50 PCR cycles). Here, virtually all classically assembled samples now exhibit nearly indistinguishable amounts of amplification on both Cre and Apo assays (188/192 replicates, 98.0%). In direct comparison, only a couple of reactions were positive for the Cre assay (X-axis; 2/192 replicates, 1.0%), whereas none were for the endogenous control gene. This impressive reduction in contamination directly results in less ambiguous data and sharply decreases the number of animals requiring reanalysis.
We repeated the above study with individual gDNA samples to more accurately mimic variability observed between individual animals. Scatterplots are again shown with routine amounts of PCR amplification (32 cycles; Fig. 4 , top row) and an excessive amount of cycling to clearly observe contamination (50 cycles; Fig. 4 , bottom row). Reactions created using traditional platforms ( Fig. 4 , left column) are contrasted to reactions assembled by an acoustic pipettor ( Fig. 4 , right column). As with the pooled gDNA sample, comparisons between the two technologies remain particularly compelling. At a routine amount of amplification (32 cycles), the ADE-assembled reactions again show no observable amplification in NTC controls (red dots, N = 192) and yield unambiguous results. Many NTC samples (138/192, 71.9%) show appreciable accumulation of signal on the X-axis [FAM fluorophore (Cre)].
Although displaying a mild increase in variability, the extreme diagnostic cycling at 50 rounds of PCR mirrors that of the 32 cycles of amplification (above) observed with the pooled sample. These individual, genetically modified mouse samples assembled by ADE show very little apparent contamination with very high cycling ( Figure 4 ): One NTC sample displays as positive for Cre detection (FAM fluorophore, 1/192, 0.5%), whereas six NTC samples display as positive for the Apo allele (HEX fluorophore, 6/192, 3.1%). No NTC reactions constructed acoustically demonstrated a detectable signal in both channels. In stark contrast, all but one NTC reaction created by a traditional laboratory robot displayed observable amplification after 50 PCR cycles (191/192, 99.5%). The vast majority of NTCs exhibited strong amplification in both channels (180/192; 93.8%) with a small subset of samples showing only Cre detection (11/192; 5.7%). The data for both 32 and 50 amplification cycles are summarized in Figure 5 .
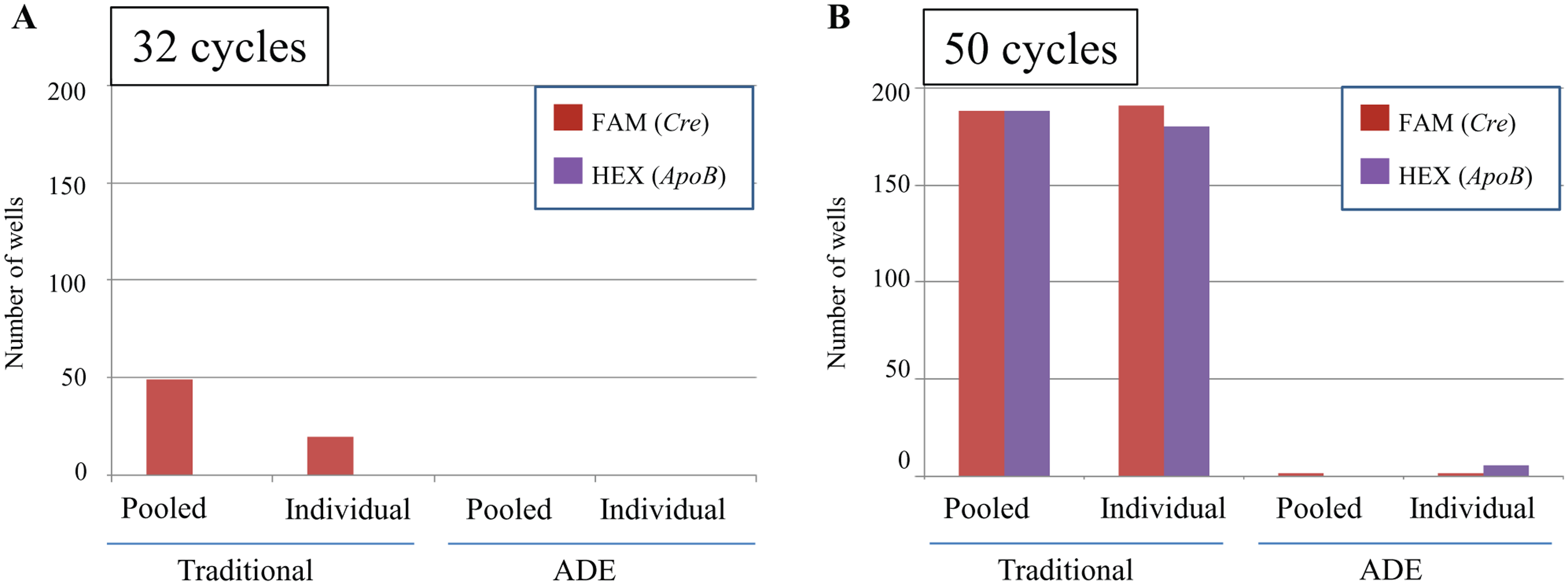
Summary of contaminated wells by assembly platform. In this illustration, a well is considered to present contamination when displaying >500 relative fluorescent units (RFUs) at the given PCR cycle value: (
As shown in Figure 4 , several of the NTC reactions directly overlap with experimental samples rendering statistical analysis impossible. In the context of a very-high-throughput genetic analysis laboratory, this can be a critical success point for a given assay or colony when this occurs at normal cycling values. Often, there is a need for all assays and protocols to follow a standard operating procedure to maximize the amount of samples that can be accurately analyzed each day. A liquid-handling system that yields less obscure data provides results in a context of a higher level of scientific integrity while simultaneously reducing the amount of reanalysis required.
Evaluation of Cross-Contamination in CE-Based Fragment Analysis
As mentioned, our laboratory uses both fragment-based PCR genotyping as well as quantitative PCR (results described above). We sought to determine whether use of ADE liquid handlers resulted in the same gains in data consistency and cross-contamination reduction. PCR reactions were assembled on both robotic platforms to interrogate PS2, a transgene known to play a role in amyloid precursor protein processing, along with IL8R, an endogenous control amplicon targeting the interleukin 8 (IL8) receptor locus. After assembly, reactions were cycled 30 times and processed on an electrophoretic capillary-based genetic analyzer. Electropherograms representing typical data observed in this cohort are illustrated in Figure 6 . Reactions were assembled by a classical pipetting robot ( Fig. 6 , top row) and an ADE device ( Fig. 6 , bottom row). gDNA from animals known to harbor the PS2 transgene were used ( Fig. 6 , left column) alongside NTC reactions ( Fig. 6 , right column).
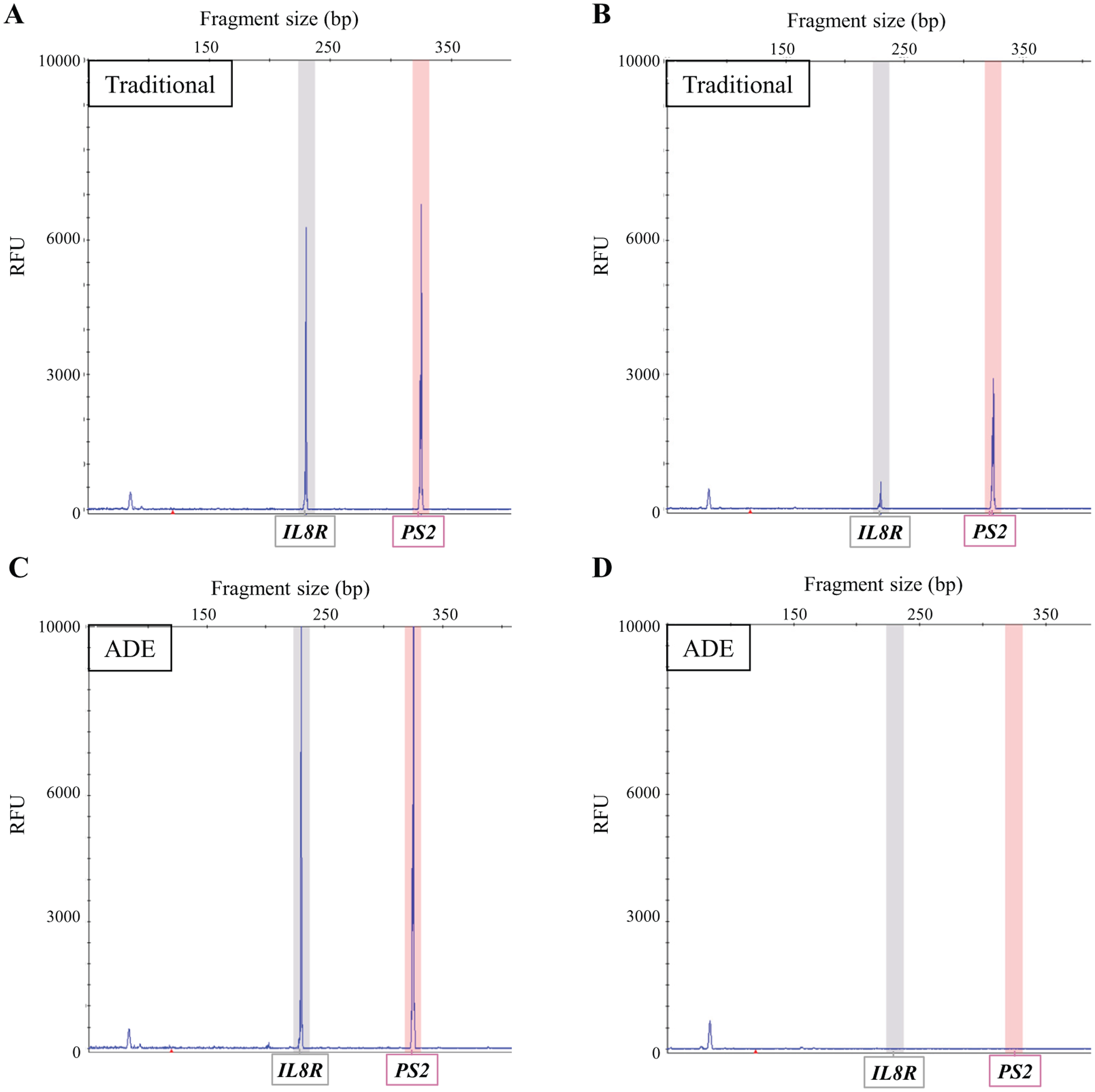
Comparison of contamination observed when performing PCR fragment-based genotyping. Electropherograms are displayed for reactions assembled on a traditional liquid handler (top row) and an acoustic pipettor (bottom row). Samples positive for (
Although the fragment peaks from the PS2+ animals ( Fig. 6a,c ) are clear, the signal intensity of the ADE-assembled PCR reactions are more intense. We observe cross-contamination results on the PCR fragment platform similarly to the qPCR platform data provided above. NTC reactions created by an aspirate-and-dispense robot illustrate ample contamination signals ( Fig. 6b ) compared to those created by an ADE robot displaying undetectable amounts of contamination ( Fig. 6d ). Notably, there is a large difference in S:N between positive and negative gDNA samples on the two assembly platforms. S:N between positive samples and NTCs is ~2.3 and ~8.3, respectively, for the PS2 and IL8R loci. In contrast, with undetectable contamination in the no-template control reactions, the S:N for positive and negative reactions assembled by ADE approaches infinity.
We assembled 96 test reactions on the traditional robotic system (48 positives, 48 NTCs) and 384 reactions on the acoustic dispenser (192 positives, 192 NTCs). Again, we found most reactions set up by the classical robotic system were significantly contaminated (44/48, 91.7%), whereas none of the 192 control reactions constructed via ADE possessed detectable contamination. It should be noted that different levels of contamination result in unique outcomes dependent on the assay. In this case, the level of observed contamination, although undesired, still results in the ability to call the genotype with a fair degree of confidence. An analyst evaluating these data could determine that there is still decent S:N between NTCs and experimental samples and conclude animal genotypes. However, working in such a mode retards analysis speed while increasing risk and errors in human interpretation.
In general, we noted that contamination of the blanks was often dependent on the last reagent pipetted before aspirating water for the “template” in an NTC reaction. Despite following manufacturer’s instructions for maintenance and operational procedures, if the last reagent pipetted was a gDNA source, we observed contaminated NTCs despite thorough tip washing. In contrast, if water was transferred previously and then water was transferred again, that NTC was often observed to have little or no detectable contamination. This suggests that, in certain cases, fixed tips combined with the use of standard washing protocols may be insufficient for particular genomics applications.
Conclusions
We have illustrated the large differences in data quality and precision that can transpire from adaptation of an ADE instrument. Moreover, the observed reduction in contamination and its effects on data interpretation and analysis can be highly impactful, especially in terms of carrying out genotyping of genetically modified animals. In addition to its precision, ADE liquid handlers clearly excel in throughput as well. All of our ADE experiments in this body of work required 10–11 minutes to cherrypick 384 gDNA samples to their corresponding assays. In contrast, these same experiments carried out on an aspirate-and-dispense platform required 64–67 minutes to complete. In our workflow, this equates a single ADE dispenser being as productive as six or seven traditional laboratory pipetting robots. This is notable considering that the classic platforms did not have the ability to spin-down plates, remove adhesive seals, or place new seals on plates; these functions were carried out by laboratory personnel and did not factor into the above time calculations. In contrast, the 10–11-minute workflow of the ADE instrument does include those steps, making the instrument much more “walk-away” than the classic robotic platforms.
ADE pipettors have been demonstrated to operate more quickly and with more precision than traditional laboratory robots while greatly reducing contamination. With such a large difference between the two platforms, we expect ADE devices to rapidly start replacing routine applications using traditional pipetting robots, such as those used to assemble PCR and qPCR reactions. Not only are the operational metrics of these devices quite advanced beyond what is commercially available, but also the return on investment (ROI) can be quite high, enabling straightforward facilitation of these devices in commercial enterprises. We took advantage of the platform to transition our genotyping reactions from 20 µl in 96-well plates to 3–5 µl reactions in 384-well plates. This equates to an annual reduction in master mix of 75% and reduction in assay reagents by roughly 50% (rather than 75% as might be expected, owning to the dead volume required in an ADE source plate).
Moreover, we were able to resolve a large resource commitment by transitioning to an ADE-based process. A given PCR assay from our inventory may be composed of 2–4 primers; a qPCR assay will contain 3–8 oligonucleotides (2–5 primers and 1–3 probes). Previously, it took a technician’s entire effort to maintain our library of ~800 genotyping assays, composed of several thousand oligonucleotide tubes. These required assay stock generation from which dilutions would be made and placed on the traditional pipetting platforms in an on-demand situation. Currently, we now order all assays premixed and aliquoted in 384-well source plates, requiring nearly no preparation to prepare for an ADE instrument. This facilitates the redirection of a technician’s efforts to more productive endeavors, such as data analysis instead of manual pipetting. Because of these reagent and personnel savings, a genomics lab could expect an ADE dispenser with accessory robotic peripherals to achieve its ROI in 1–2 years.
In addition, this body of work demonstrates the ability of ADE devices to assemble PCR and qPCR reactions with an impressive amount of contamination reduction. There are various sources of potential contamination in an automated laboratory; samples can become contaminated during DNA extraction, 31 while reaction assembly takes place, 32 or even during thermal cycling. 33 It is possible that if we had used a classical liquid handler with disposable pipette tips, rather than fixed tips, we could observe different or reduced patterns of contamination in the above experiments. In an high-throughput laboratory, however, disposable tips are often avoided. Setting up more than 1 million reactions annually could require many million disposable tips adding a significant increase to cost per genotype. Moreover, tips and tip waste occupy valuable real estate on a robot’s worksurface, decreasing a device’s per-run density. Finally, plastic tips have been shown to leach bioactive compounds into the assembled assays, affecting experimental outcomes.34–36
ADE technology has broad applications across the fields of molecular biology, genomics, and drug discovery. For instance, a workflow has been recently described that would apply next-generation sequencing (NGS) techniques to characterizing on- and off-target CRIPSR editing events in a genome. 37 Portions of such a process, notably multiplexing and barcoding steps, could be quite amenable to an acoustic-based liquid handler. Although it could be more challenging to use ADE technology for certain operations (e.g., transferring microbeads or cell suspensions), it becomes straightforward to envision replacing classical robotic systems with ones containing ADE robots for routine source-to-destination cherrypicking. In short, ADE devices could potentially carry out a large diversity of molecular biology applications, especially those processes that most benefit from rapid pipetting and a high degree of precision, or those sensitive to potential contamination sources.
Footnotes
Acknowledgements
We would like to thank additional laboratory members who contributed to this effort: Deborah Siler, Maria Martinez, Gregg Sy, and Emily Hunley. The following colleagues provided a great deal of expertise in transitioning to an acoustic-based workflow: Timothy D. Dawes, Richie Rodriguez, Peter Thana, Justin Bramwell, Randy Dyer, Howard Lee, and Mike Travis. Finally, we are thankful to Ichiro Matsumura for guidance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was completed using internal funding sources.