
Abstract
Chemoproteomics-based competition-binding assays allow the screening of compounds against endogenous proteins in cell or tissue extracts, but these assays are hampered by low throughput and high cost. Using compound pools rather than single compounds in a screening campaign holds the promise of increased efficiency and substantial cost reduction. Previous attempts to screen compounds in pools often fell short due to complex data tracking, deconvolution issues, compound interferences, and automation problems. The desire to screen compounds in a high-throughput chemoproteomics format sparked a reassessment of compound pooling. Through the integration of acoustic dispensing, we enabled a flexible pooling process, allowing mixture creation by combining randomized or specific samples to create defined pools. Automation enabled end-to-end tracking, using barcode scan check points and output files to track data and ensure integrity during the mixture creation process. The compound pooling approach proved to be highly compatible with the chemoproteomics assay technology. Pools of 10 compounds in a single well did not show compound interference effects or increased false-positive/negative rates. In the present study, four targets, TBK1, PI3Kδ, PI3Kγ, and mTOR, were screened using a chemoproteomics approach against pools of 10 compounds per well, resulting in robust hit identification.
Introduction
The chemoproteomics assay was previously introduced as a competition-binding assay based on the capturing of endogenously expressed target proteins from cell extracts by bead-immobilized nonselective capturing ligands (e.g., promiscuous kinase or HDAC inhibitors).1,2 The screen is performed as a competition-binding assay between the bead-immobilized ligands and the test compound from the screening set. Hits are obtained when a compound binds to the target protein at the same binding site as the capturing ligand and competitively inhibits target capture. The bead matrix is separated and captured proteins are eluted and quantitatively detected by spotting arrays. The assay is performed using endogenous proteins from cell or tissue extracts, which is closer to physiological conditions than recombinant systems. The chemoproteomics methodology efficiently enables the parallel screening of multiple targets in the same cell lysate, as previously demonstrated. 3 These characteristics make the chemoproteomics competition-binding assays a powerful alternative to traditional screening based on recombinant proteins and are well suited to determine the target potency and selectivity of inhibitors. The current setup enables the multiplexed screening of up to 10,000 compound wells per day.
The goal of the current study was to increase the throughput of the chemoproteomics assay platform to enable the screening of target proteins with maximized chemical diversity usually required in high-throughput screening (HTS) campaigns. HTS is a drug discovery process widely used in the pharmaceutical industry for the identification of leads and tool compounds. Ideally, HTS compound libraries cover a substantial chemical space to enable the identification of new molecular entities, but the size of the screening campaign is affected by availability of resources. 4 Several areas are impacted by this, such as compound management, reagent supplies, and the testing of the compounds in the screening assay. 5 Financial pressures, driven by the ever increasing cost of bringing a drug to market, gave rise to increased effectiveness, like miniaturization, 6 but also testing of compound mixtures instead of individual compounds (pooling).7,8 The merits of pooling in a high-throughput environment have been the subject of debate, but several groups have demonstrated applicability of this approach.9–11 Experimental errors in HTS might produce false-negative (active compound classified as inactive) and false-positive (inactive compound classified as active) results. 12 This effect could be more pronounced when screening pools of compounds due to several compound interaction effects and interferences with the assay readout. While false-positive compounds are identified as inactives in later screening phases, false-negative compounds would lead to a loss of potential lead compounds. To limit these effects, complex mixture designs have been proposed.9,13 However, the generation of assay-ready compound mixture screening plates is a challenging task, regardless if compounds are pooled randomly or a mixture design is applied. Creating compound mixture plates requires a multifaceted data tracking system, which labels pooled wells and enables the deconvolution of the pools into the individual compounds, as well as a robust liquid handling system. Despite these challenges, we considered the screening of compound pools as a practical approach to increase the capacity and economic viability of our chemoproteomics platform. Previous chemoproteomics campaigns indicate that the assays are robust, show low tendency for compound interference, and have low false-positive hit identification rates, contributing to relatively low intrinsic hit rates. However, these advantages were outweighed by reagent supply and costs, particularly for cells and detection antibodies, as well as the desire to increase the screening throughput.
Here we describe the enhancement and implementation of a novel semiautomated process for generating compound mixture plates using acoustic dispensing combined with an efficient inventory system, overcoming the logistic hurdles of generating compound mixture screening plates. We have tested and validated the approach of screening compound mixtures on the basis of a chemoproteomics competition binding assay for the protein kinase TBK1, where a screen of roughly 173,000 single compounds was followed by retesting 3345 compounds as pools of 10. This pilot gave sufficient confidence to apply the pooling approach for a campaign in which we screened 27,000 compounds in an assay for three target proteins in total: for two targets of the PI3K lipid kinase family, PI3Kδ and PI3Kγ, and for the atypical kinase mTOR. The results demonstrate that testing of pools efficiently reduced screening costs and timelines, while maintaining comparable hit rates to screens with single compounds per well.
Materials and Methods
Reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted. Compounds were dissolved in DMSO. Jurkat and Hut-78 cells were obtained from ATCC (Manassas, VA).
Creation of Pooled Plates
Pooled compound plates were created using an Echo acoustic dispenser (Labcyte Inc., Sunnyvale, CA, USA) by collapsing ten 384-well source plates into one 384-well destination plate (polypropylene V-bottom plates). Predefined volumes of 10-mM compound stock solutions (1 µL/well) were transferred from the source plates into the destination plate. This resulted in 10 µL solution per well containing 10 compounds at a final concentration of 1 mM per sample. Any well that contained fewer than 10 compounds was backfilled with DMSO to maintain the 1-mM compound concentration. Columns 6 and 18 of the destination plates were left empty for individual control addition. Each mixture contained a unique set of samples, allowing each sample to be screened only once rather than multiplexed. The Echo acoustic dispenser was controlled by an Agilent Biocel protocol (Agilent Technologies, Santa Clara, CA), which also recorded the source to destination barcode relationship. The recorded barcode file was used for upload into the inventory system (Mosaic; Titian, London, UK) for tracking the liquid handling steps and the content of the destination plate. For data tracking, a unique material identifier was generated for each compound pool. The plate map visualized in the inventory system captures the breakdown of each compound within a pooled well (
Capturing Matrix and Lysate Preparation
The capturing matrixes derivatized with immobilized ligand were prepared as described. 1 For the TBK1 chemoproteomics assay, the pan-kinase inhibitor CZC8004 and for the PI3Kδ, PI3Kγ, mTOR chemoproteomics assay, the amine functionalized analogue compounds of the inhibitors PIK93 and LY294002 were immobilized on N-hydroxysuccinimide (NHS)-activated Sepharose 4 beads (GE Healthcare, Milwaukee, WI, USA). Remaining N-hydroxysuccinimide groups on the beads were blocked with ethanolamine. The final capturing matrix for PI3Kδ, PI3Kγ, and mTOR was generated by mixing equal volumes of the PIK93 and LY294002 matrices.
Jurkat and Hut-78 cells were cultured following ATCC protocols. Cells were harvested by centrifugation and homogenized in lysis buffer (50 mM Tris/HCl [pH 7.5], 5% glycerol, 1.5 mM MgCl2, 150 mM NaCl, 20 mM NaF, 1 mM Na3VO4, 1 mM dithiothreitol [DTT], 5 M Calyculin A, 0.8% Igepal-CA630, and a protease inhibitor cocktail) using a dounce homogenizer on ice. Lysates were cleared by centrifugation at 100,000 g for 60 min at 4 °C and adjusted to 5 mg/mL total protein concentration using the Bradford assay.
Chemoproteomics Competition Binding Assay
The PI3Kδ, PI3Kγ, and mTOR chemoproteomics competition binding assay in a 384-well format using an antibody-based array readout was performed as described previously. 3 All compound dilution and addition steps were performed by an automated liquid handler (Bravo Automated workstation; Agilent Technologies). Then, 0.25 mg extract of Hut-78 cells and 2.5 µL of capturing matrix per well were incubated in the presence of test compounds or controls at 4 °C. DMSO concentration was 2% (v/v). Each plate contained 16 positive (100 µM Piramed PI-103) and 16 negative (2% v/v DMSO) controls. After 2 h incubation, the nonbound fraction was removed by washing the beads with lysis buffer (50 mM Tris-HCl, 0.4% [v/v] Igepal-CA630, 5% [v/v] glycerol, 150 mM NaCl, 1.5 mM MgCl2, 25 mM NaF, 1 mM sodium vanadate, 1 mM DTT, complete EDTA-free protease inhibitor tablet [Roche, Basel, Switzerland], pH 7.5). Proteins retained on the beads were eluted in sodium dodecyl sulfate (SDS) sample buffer (100 mM Tris [pH 7.4], 4% [w/v] SDS, 2 0% [v/v] glycerol, 0.01% [w/v] bromophenol blue, 50 mM DTT) and spotted onto three nitrocellulose membranes as identical copies (400 nL/spot) using an automated pin-tool liquid transfer (Biomek FX; Beckman Coulter, Brea, CA). After drying, the membranes were rehydrated in 20% (v/v) ethanol and processed for detection with three specific primary antibodies, respectively: anti-PI3Kδ (sc-7176, 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-PI3Kγ (ABD-026L, 1:100; Jena Bioscience, Jena, Germany), and anti-mTOR (#2972, 1:500; Cell Signaling Technology, Beverly, MA) in the presence of 0.4% Tween followed by incubation with an IRDye 800–labeled secondary antibody for visualization (anti–rabbit 926-32211, anti–mouse 926-32210; LiCOR, Lincoln, NE [1:3000]) in the presence of 0.2% Tween. Spot intensities were quantified using a LiCOR Odyssey scanner, and percentage inhibition was calculated using positive and negative controls as 100% and 0% inhibition, respectively.
The TBK1 assay was done in analogy to the PI3Kδ, PI3Kγ, and mTOR assay with the following exceptions: 0.125 µg extract of Jurkat cells and 1.25 µL CZC8004-inhibitor matrix (1 mM coupling density) per well were applied. Then, 20 µM staurosporine was used as a positive control. For detection, an anti-TBK1 primary antibody (04-856, 1:6000; Merck Millipore, Billerica, MA, USA) and an IRDye 800–labeled secondary antibody (anti–rabbit 926-32211; LiCOR [1:5000]) were used under the same conditions as above.
For testing compounds in a dose-dependent manner, the maximum assay concentration was 200 µM, and 1:3 dilution steps for a total of 11 data points were applied. Odyssey raw data were uploaded and analyzed in ActivityBase (IDBS, London, UK). Concentration-response curves were fitted using a four-parameter logistic fitting module in ActivityBase using the following equation:
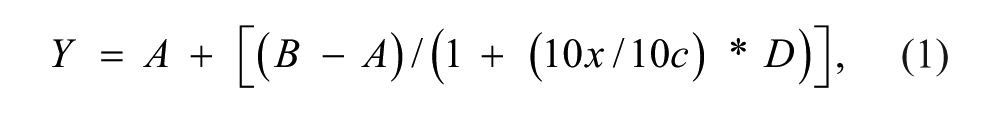
where A = minimum response, B = maximum response, c = log(IC50), D = slope factor, and x = log(Molar compound concentration). pIC50 values equal –c in the above equation and are the negative logarithm of the IC50.
Estimated IC50 values were determined on the basis of single concentration inhibition values using the following formula:
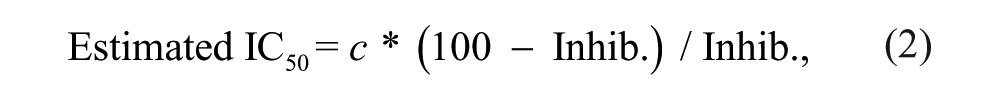
where c = screening concentration (µM), and Inhib. = inhibition at screening concentration (µM). pIC50 values are the negative logarithm of the IC50.
Results
Here we describe a high-throughput chemoproteomics-based screening platform to enable the multiplexed screening of small-molecule libraries as pools of 10 compounds per well (
Fig. 1
). The assay was performed in a competitive binding mode, and active compounds were identified by the reduced binding of the target protein to the capturing beads derivatized with selected ligands.
3
After capturing the target proteins in the presence of test compounds, the capturing matrix was washed to remove unbound protein as well as the test compound. The captured proteins were eluted under denaturing conditions and spotted in nanoliter volumes on nitrocellulose membranes in multiple copies followed by antibody-based detection of the target proteins. The assay was performed in a 384-well format, and the liquid handling steps were automated using liquid handling robots equipped with pipette tips or a pin-tool for the transfer of eluate on the nitrocellulose membranes. Screening campaigns of pooled samples comprised several process steps (
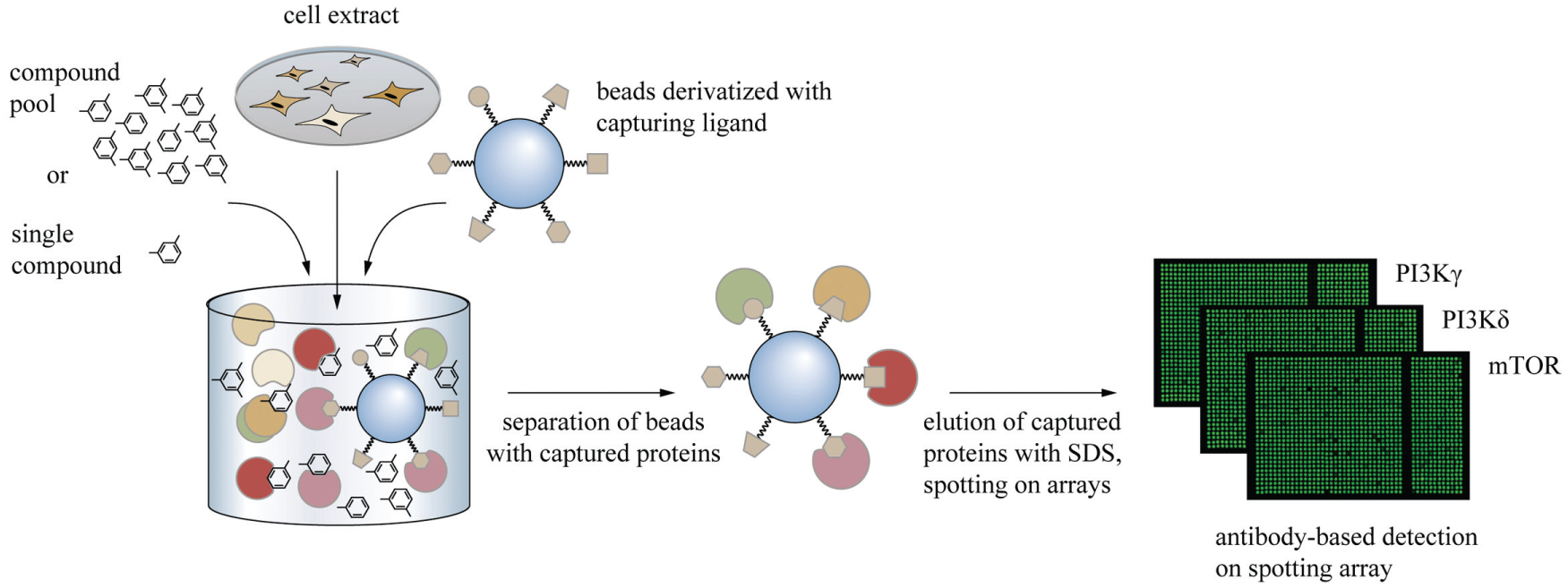
Chemoproteomics competition-binding assay using antibody-based detection. Beads derivatized with capturing ligands are applied for competition binding assays. Antibody-based detection enables multiplexed screening of up to 10,000 compound wells per day in cell extracts.
Generation of Compound Pools
Using an adaptive pooling strategy, samples were combined by collapsing up to 10 plates into one, and compound pool plates were generated using an acoustic dispenser. Then, 1 µL of up to ten 10-mM compound stock solutions per well were combined, yielding a 10-µL solution with each compound at a final concentration of 1 mM. If the pool contained fewer than 10 compounds, DMSO was added to keep the final concentration and the volume consistent. Exploitation of acoustic dispenser capabilities enabled the creation of pools in small volumes, which allowed for significant reduction in compound and solvent consumption in comparison to other liquid handling systems. Furthermore, the new approach was highly efficient, enabling the physical creation of pooled plates for the 27,212 compound set to be completed within 3 working days. All liquid handling steps were controlled and tracked by the Agilent BioCel automation platform, enabling a complete recording of the source to destination plate relationship. The acoustic dispenser was integrated into the automation platform by a custom-made program, which allowed walk-away time while processing. The inventory system was configured to include a module that allowed the creation and tracking of pool generations as well as the deconvolution of the pools into its individual compounds. This pilot method still has its limitations. While the physical generation of pool plates and deconvolution singleton plates was automated, the electronic plate creation was a manual process. For each pooled plate, a query within the inventory system was run to generate an electronic plate map. To determine the exact number of compounds within a well, a manual calculation was necessary using the electronic plate map as a guide. It is planned that both the inventory and automation systems will be altered to avoid these time-consuming and potentially error-prone manual steps in future. The acoustic dispenser controlled by the automated platform ensured efficient generation of compound pool plates, which can be directly used in multiple chemoproteomics screens. This setup ensures complete tracking of each liquid handling step and enables a reliable and simple deconvolution of the compound pools, which finally appear active in the screen.
Chemoproteomics Screen for TBK1: Comparison of Compound Pool with Single-Compound Screening
To assess the feasibility of screening compound pools, we selected 3345 compounds from a previous TBK1 chemoproteomics screening campaign and retested them as pools of 10 compounds per well (352 pools). In the initial TBK1 screen, diverse and kinase-focused library sets were tested, in total 172,961 compounds, at a final assay concentration of 20 µM. The assay was robust, and the data quality was high (
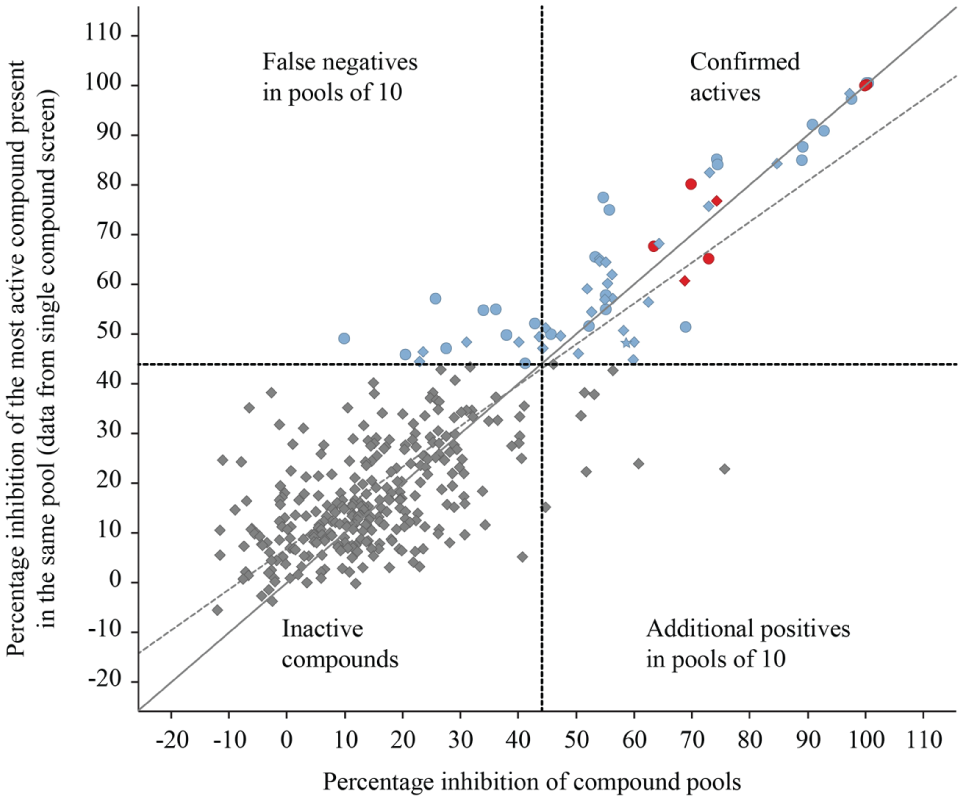
In total, 77% of the active compounds identified in a TBK1 single-compound screen were reidentified in the compound pools. Correlation between percentage inhibition of the compounds pools and the percentage inhibition of the most potent compound of each pool. The most potent compounds of the false-negative pools showed inhibition values close to the hit cutoff in the initial screen (less than 60% inhibition). The shape indicates the results of the compounds in the dose-dependent analysis of the initial screen: circles: active compounds; star: inactive compound; diamonds: not retested in a dose-dependent manner. The colors indicate the number of compounds per pool, which were active in the TBK1 single-concentration screen; blue: 1 compound, red: 2 compounds; gray: no active compound. The solid line represents equality and the dotted line the fit (R2 > 0.7). The dashed line represents the hit limit at 44% inhibition.
Pools with an inhibition below 44% that contained compounds that appeared active in the initial screen are considered false negative. However, the active compounds in the false-negative pools were of low potency with an inhibition value of at most 60% in the initial screen. All pools containing compounds that showed in the initial screen an inhibition larger than 60% appeared active, demonstrating that highly potent compounds were correctly identified from the pools in all instances.
Nine compound pools showed an inhibition above 44%, despite the fact that they did not contain any active compound based on the results of the initial screen and were considered likely false positives. Potential false positives would be identified in follow-up screening processes, and a potential false-positive rate in this range was deemed acceptable. In summary, the results of the feasibility study based on the TBK1 chemoproteomics assay indicated that screening of pools represented a feasible extension of the chemoproteomics screening format.
Multiplexed Chemoproteomics Lipid Kinase Screen on Compound Mixtures
Next, we screened a diverse library set comprising 27,212 compounds as pools of 10 per well in a high-throughput chemoproteomics binding assay for three targets. In this screen, two members of the PI3K lipid kinase family, PI3Kγ and PI3Kδ, and the atypical kinase mTOR were addressed. The final assay concentration of the individual compounds was 20 µM, respectively, leading to a total compound concentration of 200 µM per well. The signal-to-background ratios were 18 for PI3Kδ and 3 for PI3Kγ and mTOR, respectively, and the Z′ factors between 0.8 and 0.9 indicated a high data quality for the three targets (
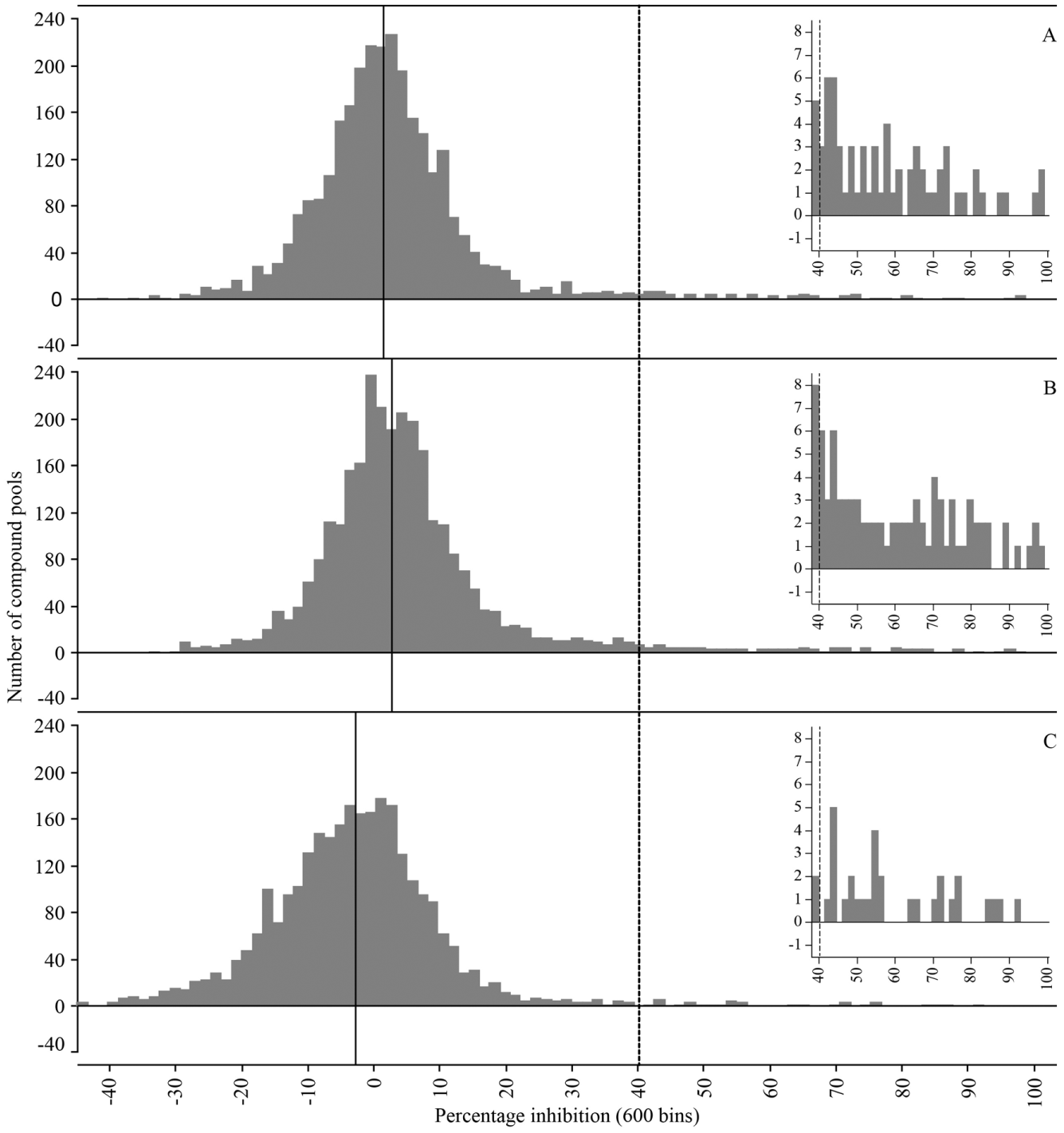
Identification of 126 active pools in the PI3Kδ, PI3Kγ, and mTOR screen from a 27,212-compound library as pools of 10 per well. Distribution of reduced bead binding by compound pools (% inhibition) for PI3Kδ (
One single compound pool showed very high signal intensity and therefore resulted in inhibition values below −100% for all three targets. This might have been due to one or more compounds within the pool interfering with the assay readout (
The hit limit was set to 40% inhibition at a 20-µM final screening concentration, leading to the identification of 62, 76, and 30 active compound pools for PI3Kδ, PI3Kγ, and mTOR, respectively. In total, 126 pools appeared active for one or more target proteins. For the deconvolution of the compound pools showing more than 40% inhibition, 1226 compounds have been tested as a single compound per well in the PI3Kδ, PI3Kγ, mTOR assay at a final assay concentration of 20 µM. Thirty-one compounds were not included due to limited availability. Most of the inhibition values of compound pools were confirmed, evidenced by plotting the inhibition value of the pools against the inhibition value of the most active compound of the pool ( Fig. 4 ). Pools were considered confirmed if the inhibition values of the pool and most active compound of the pool deviated by less than 16%, which corresponds to a ±0.3 log unit shift of estimated pIC50 value. The confirmation rate of the active pools ranged from 57% to 81% for the kinases ( Fig. 4D ). Four compound pools showed inhibition values above 55% for all three targets but were not confirmed for any of the targets. False positives could originate from errors at any stage of the assay procedure and might be independent of screening individual compounds or compound pools. For several of these pools, not all compounds could be retested due to depletion of the stock solution, which limited the confirmation of active pools. Most of the other not confirmed pools had inhibition values closer to the hit cutoff in the original screen. As for the TBK1 chemoproteomics screen, we considered the false-positive rate of the compound pools as acceptable, as these were easily identified by retesting the individual compounds at single concentration.
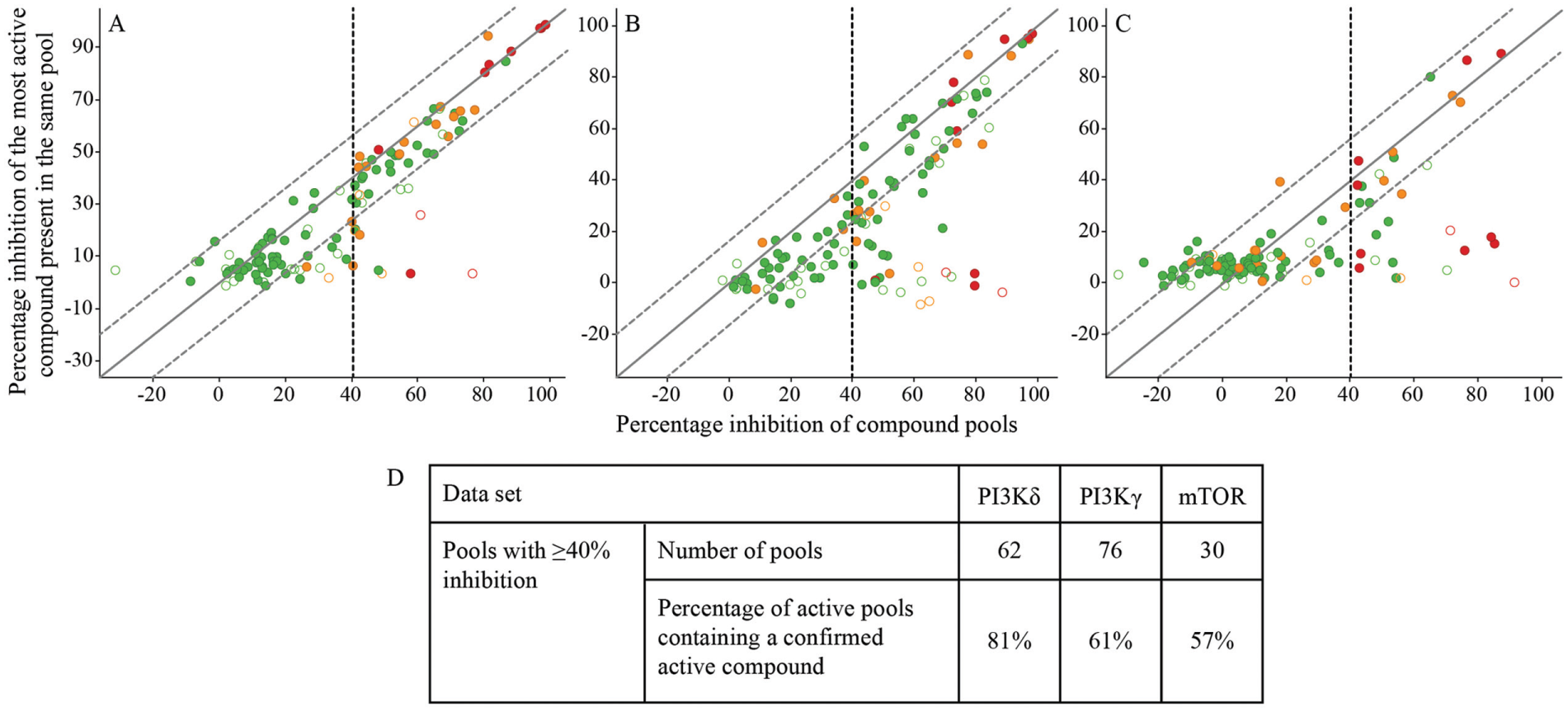
Between 57% and 81% of the active pools were confirmed by retesting the individual compounds in the PI3Kδ, PI3Kγ, and mTOR chemoproteomics assay. Correlation between percentage inhibition of the compounds pools and the percentage inhibition of the most potent compound of each pool. Filled circles display pools, of which all compounds were retested individually, and open circles display pools, of which not all compounds were retested. The colors indicate the number of targets, for which the pool showed more than 40% inhibition: green: one target; orange: two targets; red: all three targets. The solid line represents equality and the dashed lines ±16% of equality as confirmation cutoff. (
Confirmation of Active Compounds
All compounds that appeared active in the deconvolution step for any of the three targets, PI3Kδ, PI3Kγ, and mTOR, were retested in the same chemoproteomics assay in a dose-dependent fashion as replicates. Fifty-nine, 64, and 22 hits were identified for PI3Kδ, PI3Kγ, and mTOR, respectively, with a pIC50 range of 3.8 to 6.8, yielding an overall hit rate of 0.1% to 0.2% (
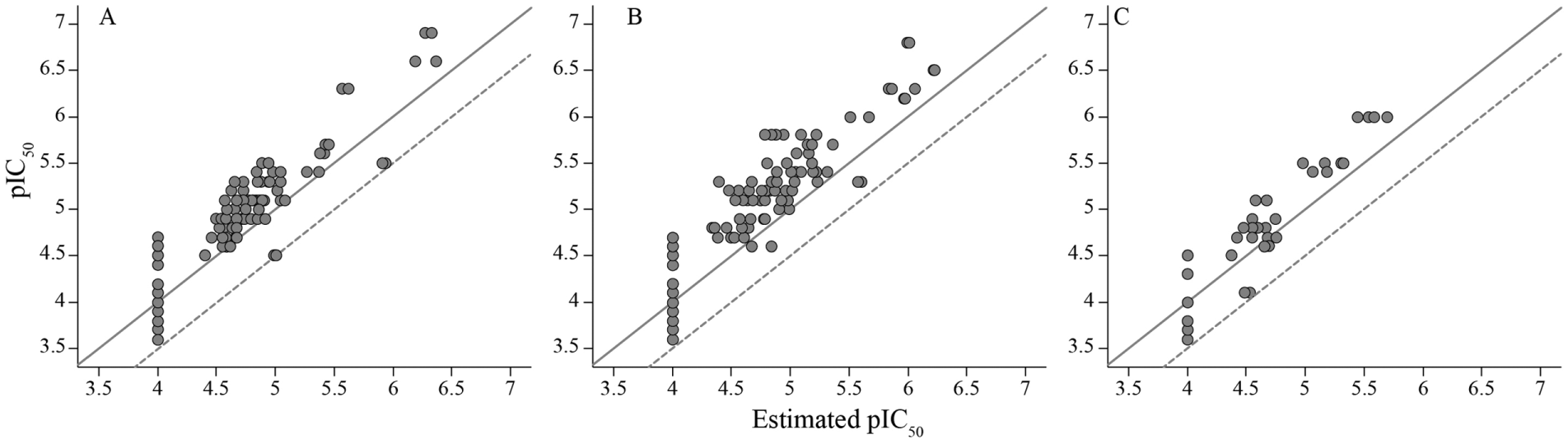
All active compounds from the deconvolution screen were found active in the dose-response confirmation step performed for PI3Kδ (
In multiplexed screening campaigns, the potency of the test compounds enables one to derive the selectivity windows for the individual target proteins ( Fig. 6 ).
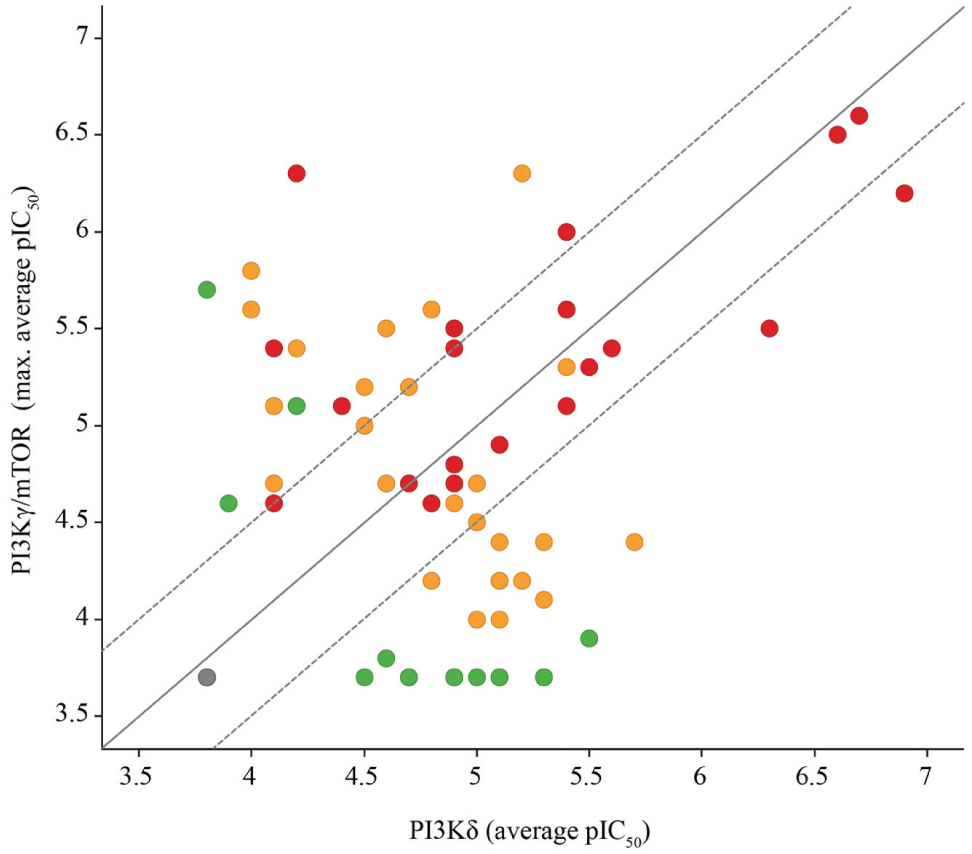
Multiplexed chemoproteomics screening enables direct determination of selectivity of screening hits. Active compounds of the deconvolution screen were retested in a dose-response mode in replicates for PI3Kδ, PI3Kγ, and mTOR. This enables the determination of selectivity windows for the three target proteins. The colors indicate the number of targets, for which the compound showed a pIC50 equal to or larger than 4: green: one target; orange: two targets; red: all three targets. The solid line displays selectivity and dashed lines a selectivity window of 0.5 log units. Values with a mean pIC50 below 3.7 were set to 3.7.
The example demonstrates that the most potent hit compounds were active for all three targets and not selective for PI3Kδ. However, some of the PI3Kδ hit compounds showed a selectivity window of at least 0.5 log units and were active against only one of the two other targets.
Discussion
In this study, we confirmed that our previously introduced chemoproteomics assays 3 are in principle compatible with HTS campaigns. The increase in throughput was enabled by the multiplexed screening of pools of compounds for several target proteins in parallel. In the first example, the TBK1 screen, 172,961 compounds were screened in more than 202,000 compound wells using a one-well/one-compound approach, resulting in a hit-rate of 0.8%. The low hit rate and the robust assay quality would enable one to test also pools of 10 compounds per well. This would substantially increase the screening capacity, suggesting that in principle, a 2 million compound screen in a chemoproteomics competition-binding assay would be feasible using an adaptive compound pooling strategy. When screening compound pools instead of single compounds per well, the saving of time and resources has to be balanced against potential disadvantages such as increased false-positive or false-negative rates. Experience gathered in previous screening campaigns based on the chemoproteomics assay showed that hit rates were typically below 1.0 %, depending on the nature of the targets and the compound concentration used. We suggest that the low hit rates in these screens are due to low false-positive rates since a binding assay in a cell extract with high protein content in a high-salt/high-detergent buffer should discriminate against many of the effects known to cause false positives.14,15 Pooling 10 compounds per well represented the best compromise between a substantial gain in throughput, on one hand, and managing a high hit rate, total concentration of small molecules, and DMSO, on the other hand. The assay has a very low liability to compound interferences with the assay readout for multiple reasons. On one hand, the largest part of the compound is removed, when the bead matrix is separated from the unbound protein. On the other hand, the secondary antibody used to read out the spotting array is coupled to a dye fluorescing at infrared wavelengths. This leads to less compound interference effects than the dyes typically used in standard biochemical fluorescence assays. The low hit rate and low incidence of false positives makes this an ideal approach for screening pools of compounds.
The distribution of inhibition values of the pooled screens centered on zero and was similar to previous screens on single compounds using the same assay platform. There was no indication of significant compound interferences or other effects due to the overall high compound concentration. We identified one compound pool, which showed a very high fluorescent signal for all three targets of the PI3Kδ, PI3Kγ, and mTOR screen. This pool might have contained one or more compounds that, interfering with the assay readout, could have overshadowed potentially active compounds. To reduce this risk, all compounds of pools with high fluorescence should be included in the deconvolution screen.
The false-positive rate varied in the chemoproteomics assays for the four target proteins. TBK1 or PI3Kδ showed a higher confirmation rate than PI3Kγ and mTOR.
The low confirmation rate for mTOR could be partially explained by the higher impact that false positives have when the hit rate is low. For TBK1 and PI3Kδ, the assay robustness was higher compared with PI3Kγ and mTOR, as the signal-to-background ratios and the distribution of the inhibition values indicated. Differences in the robustness of the chemoproteomics screening assays for the individual targets can be related to the expression level of the protein, the affinity to the immobilized ligand, and the quality and affinity of the detection antibody.
Besides a normal assay variation, which occurs also when screening single compounds per well, false positives could be generated by a cumulative effect of multiple weakly active compounds in a pool, by target modification, interference in the assay readout, and promiscuous aggregate-based inhibition. 16 In our strategy, false-positive pools were efficiently discriminated from true actives in the deconvolution screen, where all compounds of the active pools have been retested individually at a single concentration, as demonstrated for the PI3Kδ, PI3Kγ, and mTOR screen.
False negatives, however, are more of a concern, as potential lead compounds might be missed. False negatives in screens of pooled compounds could be caused by limited solubility at the overall high final assay concentration, which was 10-fold higher than the concentration of each individual compound. Furthermore, false negatives may occur due to assay interferences of one of the compounds within a pool, which might overshadow the effect of an active compound. In the case of TBK1, an approximate rate of false-negative compound pools could be deducted by comparing the screen of individual compounds with the screen of the same compounds as pools of 10. Seventy-seven percent of the pools containing active compounds were confirmed by this approach, and the missed hits were close to the hit cutoff of 44% inhibition. Compounds, which showed an inhibition above 60% in the initial screen, had a confirmation rate of 100%. This suggests that compounds of high potency are less prone to appear as false negative. An option would be to further lower the hit limit to a 2-fold standard deviation of the mean to further decrease chances of false negatives. In case of the PI3Kδ, PI3Kγ, and mTOR screen, 196 pools would have been selected for deconvolution at a hit cutoff of 30% inhibition. In that case, around 2000 compounds would have been retested individually at a single concentration, which would have been still feasible.
Matrix-based compound pooling and deconvolution approaches have been published, in which all compounds are assayed more than once, each time with a completely different set of pools. 17 These approaches make redundant the need of a deconvolution step of active compound pools and has the potential to decrease false-negative rates. 9 However, matrix-based pooling would require the presence of each compound in multiple pools, resulting in substantially higher usage of reagents that would not have been economical for a chemoproteomics screen. Due to the usually low hit rate in the chemoproteomics screens, testing compound pools in the initial screen only once, followed by a deconvolution screen, resulted in the best strategy. The efficiency of this approach was demonstrated by the good correlation of the estimated pIC50 values determined at single concentration in the deconvolution screen with the average pIC50 value determined in dose-response experiments.
A challenge of screening compound pools was achieving not only the required robustness of the assay system itself but also the compound management steps. Therefore, we developed a compound pooling strategy to create compound pool plates for HTS in a semiautomated environment while ensuring data integrity. By programming the Agilent Biocel, pooled plate creation was automated using an integrated acoustic dispenser to transfer single-well compounds into pooled wells while tracking the source to destination relationship. While there are other liquid handlers capable of faster low-volume dispensing, the use of the acoustic dispenser also allowed for noncontact, wash-free dispense, reducing plate processing time and cost. 18 The overall cost reduction for compounds used was 80%. Compared with the use of an eight-span liquid handler, 11 liters DMSO were saved for the 27,212 compounds by not needing any wash cycles in between the pipetting steps. Furthermore, generating plates with an acoustic dispenser in contrast to an eight-span liquid handling system reduces overall timelines. The creation of the pooled compound set of 27,212 compounds using the acoustic dispenser reduced the process time by 88%. A common limitation of compound pooling is the tracking of the liquid handling steps as well as storing the information of the plate content. To overcome this hurdle, the inventory system was configured to allow the electronic creation and tracking of pooled wells. It also allowed an efficient way for the deconvolution of pools into the individual compounds. This was required in particular when creating the plates of the individual compounds of the active pools in the deconvolution screen. By combining data tracking and automation, a simplified process was developed, allowing for mixture creation at the time of request while optimizing the amount of sample used, ensuring data integrity, and reducing the time spent plating compound pools.
We calculated that screening large compound libraries in pools of 10 compounds per well followed by a deconvolution screen prior to hit conformation in a dose-dependent fashion can reduce screening costs up to 8-fold and accelerate screening timelines substantially. This approach enables the screening of larger numbers of compounds in assays with limited capacity.
Both the reduction of assay costs and time as well as the option to screen larger library sets might also be beneficial for cell-based or phenotypic assays. Prerequisite is a robust assay system with an intrinsic low hit rate and little inclination to compound interferences.
In conclusion, we have provided evidence that screening pools of compounds in a robust assay system, such as the chemoproteomics competition-binding assay, enables the identification of active molecules, which can then serve as starting points for further development of probes and drugs. We developed an efficient pooling strategy using the acoustic dispenser coupled to an inventory system, which enables efficient screening of large compound sets in expensive or time-consuming setups. Increasing the number and therefore the chemical diversity of the tested compounds can ultimately affect the successful outcome of the screening campaign. In addition to target-based approaches, other assays with limited throughput, such as phenotypic assays and affinity selection mass spectrometry, may equally benefit from screening pooled compound libraries.
Footnotes
Acknowledgements
We thank John Russell, Joyce Ebrahiem, and Heather Walters for their contribution to developing and handling the creation of the pooled compound plates; Jana Krause, Kira Weis, and Andrea Wolf for supporting the processing of the chemoproteomics assays; Juergen Stuhlfauth for cell culture and lysate preparation; Frank Weisbrodt for help with the figures; and Sue Crimmin and Gitte Neubauer for discussions and support.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.