
Abstract
Vesicles represent an important class of nanoscale drug delivery vehicles. To significantly reduce the time and resources that are required to optimize these drug carriers, this review article discusses the mathematical models that have been derived for understanding the formation of vesicles and their stability, as well as for predicting drug loading and their release. With regard to vesicle formation and stability, the packing parameter can be used to predict how the solution environment, surfactant composition, and surfactant molecular architecture can influence the supermolecular self-assembled structures that are formed from amphiphiles. In the context of drug delivery, this is useful for facilitating vesicle formation and stability during transit through the body. At the target site, this information can be used to help trigger a rapid release of the drug. With regard to drug loading, kinetic and equilibrium models provide guidelines for appropriate pH conditions and drug incubation times during loading. The diffusivity, partition coefficient, and bilayer thickness also play significant roles during loading and release of the drug. Our hope is that more researchers in this exciting field will complement their experimental approaches with these mathematical models to more efficiently develop vesicle-based drug carriers.
Overview
Vesicles are spherical aggregates composed of an aqueous core enclosed by a bilayer membrane formed from the self-assembly of amphiphilic molecules. Vesicles composed of lipids (i.e., liposomes) were first described by Dr. Alec D. Bangham 1 in 1964 after adding a negative stain to dry phospholipids and observing the system through an electron microscope. A little more than three decades later, the term polymersome was coined 2 for vesicles composed of polymers, and applications involving both liposomes and polymersomes have grown dramatically in recent years. 3 A branch of polymersomes has emerged more recently, which uses amino acids as the basic polymer building block, termed polypeptide vesicles. 4 Developments in academia and industry for characterizing these systems have led to the success of vesicles in the field of drug delivery. Currently, one of the most well-established commercial vesicle-based drug delivery systems is Doxil, which is used to treat cancer. This formulation consists of liposomes coated with an outer layer of polyethylene glycol (PEG) that encapsulates the chemotherapeutic doxorubicin. Vesicle systems loaded with other small-molecule drugs, such as Myocet and Ambisome, are being produced commercially. 5 Small molecules are not the only cargo option for these carriers, as vesicles have been investigated for their ability to deliver proteins and other types of biomacromolecules. 3
A significant amount of time and resources is required to develop vesicle systems into effective drug delivery vehicles. Mathematical modeling can therefore be used to aid in the optimization process and reduce the amount of time and resources that are required. This review represents part one of two articles that highlight the mathematical models that can be used to help identify design criteria for engineering vesicle systems. The two review articles together address vesicle drug delivery starting from the molecular length scale and ending at the whole-body length scale. This review article in particular focuses on the molecular and vesicular length scales, as it discusses mathematical models for vesicle formation and stability along with drug loading and release. The follow-up review article considers targeted vesicle interactions with cells, tumors, and the entire body. It is our hope that our review of the mathematical models will provide additional tools to assist investigators in developing vesicle-based drug delivery systems, which in turn will further the fascinating and promising field of vesicle-based drug delivery.
Vesicle Formation and Stability
Introduction
The success of vesicles as drug delivery vehicles is, in part, due to understanding the mechanisms and principles of vesicle formation and stability. One of the most commonly used models for vesicle formation is the molecular packing parameter, P, which is a simple but useful equation. This dimensionless parameter is given by the following equation:

where v is the volume occupied by the hydrophobic portion of the surfactant, l c is the critical chain length of the hydrophobic portion, and a o is the representative surface area occupied by the surfactant. The value of P can be used to predict the supermolecular self-assembled structures of a surfactant solution ( Fig. 1 ). 6 This molecular perspective of the geometric packing is related to parameters of the macroscopic aggregate structural geometry by 7
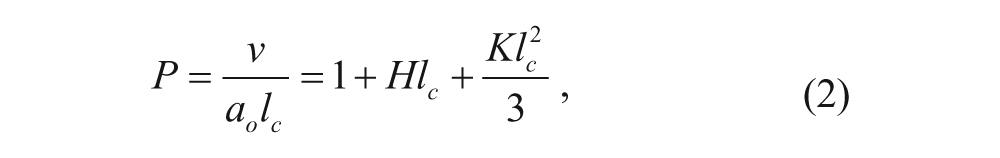
where H is the mean curvature and K is the Gaussian curvature.
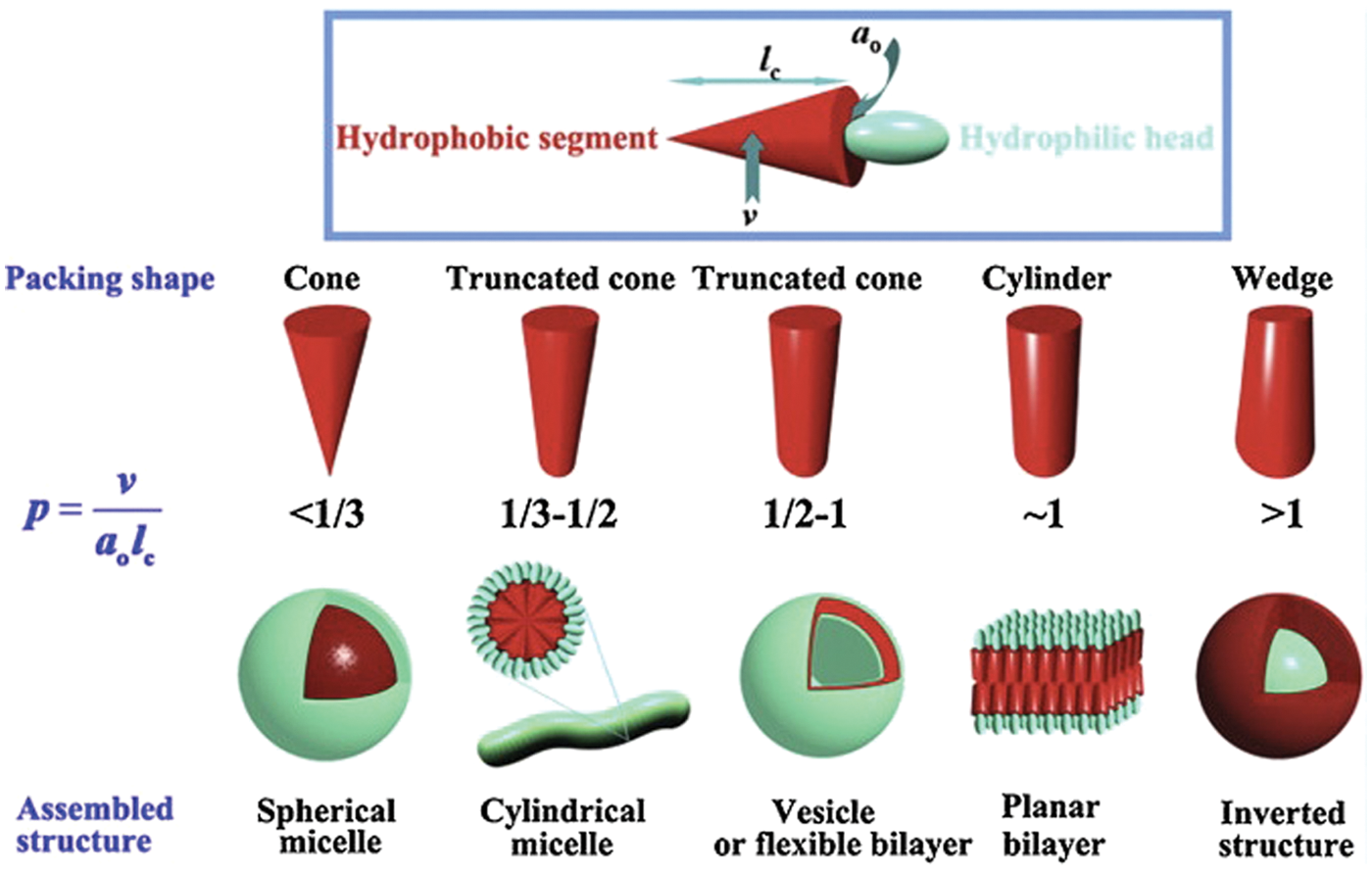
Graphic representation of individual surfactant geometry and the corresponding aggregate structure. (Li 2011). Reprinted from Progress in Polymer Science, 37, Zhang, J., Li, X., Li, X., Stimuli-triggered structural engineering of synthetic and biological polymeric assemblies, 1130-1176, 2012, with permission from Elsevier.”
A thorough understanding of the geometry and thermodynamics behind the packing parameter is important to develop a strong grasp of vesicle formation, which can be used to identify design criteria for engineering vesicular drug delivery systems. Prior to discussing the packing parameter, we will first give a brief background of the vesicle formation process.
Vesicle Formation Mechanisms
There are many reviews that describe the mechanisms and methods of liposome8,9 and polymersome10–12 formation, both in general and with respect to drug delivery. The techniques used to create liposomes and polymersomes are very similar. Here, we will briefly summarize some of the methods and concepts involved in vesicle formation.
Although there are many different procedures for forming vesicles, the mechanisms by which vesicle structures form can be organized into two general categories, which we will refer to as film hydration and spontaneous aggregation. In film hydration, dry lamellar layers of amphiphilic molecules (lipids or block copolymers), which had been previously crystalized to form a flat film, are rehydrated with an aqueous solvent. During this process, the top layers of the film start blistering with local swelling due to hydration and eventually grow to form bulbs. These techniques typically require an energy input, usually in the form of sonication. Upon agitation, the bulbs shear off and close, resulting in the formation of bilayer vesicles. The second general mechanism involves a morphological transition of the aggregates from micelles to bilayer vesicles. This phenomenon is dependent on the critical micelle concentration, which can be conceptually described as the surfactant concentration at which any additional surfactant will form micelle aggregates rather than partitioning to an interface between hydrophobic and hydrophilic phases.13,14 There are several different methods that use this process, but they all result in the initial aggregation of surfactant into spherical micelles followed by a transition to cylindrical micelles and then to disklike micelles. These disks tend to aggregate with each other and grow to reduce the interfacial area between the disk belt and the aqueous solvent. Once the disks reach a critical radius, they will start to bend and eventually close upon themselves to stabilize as liposomes. The critical radius size is dictated by the competition between closing the vesicle to reduce the exposure of the hydrophobic tails to solvent and resisting the bending of the bilayer membrane to form the vesicle. During these formation processes, the molecular packing parameter is an important tool that can be used to predict equilibrium supermolecular structures.
Packing Parameter as a Geometric Constraint
Much effort has been put into characterizing and rationalizing surfactant self-assembly. Israelachvili and coworkers in 1976, 15 following the approach of Tanford, 16 generated a framework that tied together thermodynamics, interaction-free energies, and geometric constraints. This was the beginning of the molecular packing parameter, equation 1, which has been widely cited in chemistry, physics, biology, and engineering for the past 35 years.
It has been shown that specific aggregate shapes, such as spherical micelles, cylindrical micelles, and bilayer vesicles, correspond to specific values of the packing parameter. Specifically, the packing parameter values are derived from geometric arguments, and they have reasonably predicted experimental data. 17
Given that R is the radius of a spherical or cylindrical micelle and N is the number of surfactant molecules in one micelle (i.e., the aggregation number), we can derive surface area and volume equations in terms of R and N. For spherical micelles in an aqueous solution, we have the following equation for the surface area:

Rearranging to solve for the aggregation number yields

Similarly, we can relate N to the spherical volume as follows:

We can again solve for the aggregation number, and equate it with equation 4 to yield
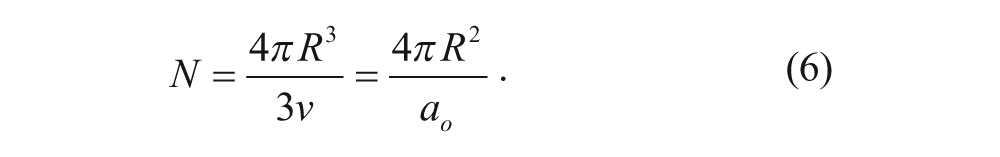
Rearranging yields an expression that is similar to the packing parameter.

Because R will always be slightly shorter than the fully extended critical chain length l c , we can substitute the inequality to yield

for the packing parameter value of spherical micelles. This results in individual surfactant molecules occupying space in the shape of shallow cones, which can be seen in Figure 1 .
Using analogous arguments for cylindrical micelles, where L is the length of the cylinder, we have the following equation for the cylindrical surface area, equation 9, and volume, equation 10:


Combining and rearranging, and taking note that R < lc, we have the following:

Because surfactant systems transition from spherical to cylindrical micelles as the packing parameter value increases, we can plug in the spherical micelle inequality, equation 8, to arrive at

for cylindrical micelles. It should be noted that the number of surfactant molecules in the end caps is typically much smaller than the number of surfactant molecules present in the rest of the cylinder; therefore, end effects can be neglected. The shape occupied by the individual surfactant molecules in a cylindrical micelle is described as a truncated cone.
Deriving the expression for the packing parameter of a vesicle follows a different approach. First, a flat bilayer is considered. Because there is no curvature for this self-assembled supermolecular structure, the surface area occupied per molecule on the top and bottom are equal, giving the occupied space a cylindrical or rectangular prism shape with a volume equation


Because the vesicle morphology is an intermediate between a flat bilayer and a cylindrical micelle, the packing parameter values for a vesicle are given by

for vesicles. Because the packing parameter does a reasonable job of predicting and explaining experimental data, 17 it is important for researchers to understand the principles that govern the variables of the packing parameter to have full control over their vesicle delivery system.
Thermodynamic Manipulation of the Packing Parameter
From a practical standpoint, the area per molecule a o is generally considered the most influential variable of the packing parameter and can be defined as the total surface area of the aggregate divided by the number of surfactants in the aggregate (or the number of surfactant per leaflet for bilayer aggregates). This value is commonly mistaken as a simple variable that is dictated solely by the geometry of the hydrophilic region. Although the molecular architecture is certainly influential, a o is an equilibrium value based on minimization of the free energy resulting from interactions between neighboring surfactant head groups. For the purpose of this review, we will use the original framework developed by Tanford 16 for our explanations. However, it should be noted that his work has been further developed into more complex models. 18
Tanford developed a model for the change in free energy when a single surfactant molecule is transported from an infinitely dilute solution of water to the core of an aggregate. After dividing this free energy change by the thermal energy (kBT), it is given by
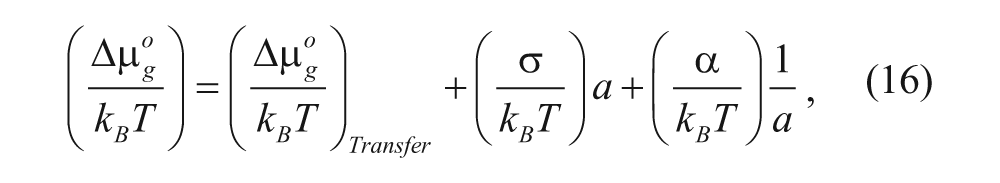
where σ is the interfacial free energy per unit area, α is the head group repulsion parameter, k B is the Boltzmann constant, T is the absolute temperature, and the other terms will be described below. This equation has three terms that contribute to the formation and stabilization of surfactant aggregates, where negative and positive free energy values correspond to favorable and unfavorable contributions, respectively. The first term is the free energy contribution from the transfer of the hydrophobic portion of the amphiphile from an unfavorable aqueous environment to the more desirable hydrophobic core of the aggregate and therefore will have a negative value in the context of vesicle formation. This transfer phenomenon is driven by the high entropic cost of creating a solvation shell of H2O molecules around the hydrophilic segment when it is in solution 17 and therefore favors surfactant aggregation. Groups have used this first term to control the initiation of vesicle formation. For example, Armes and coworkers 19 used a pH-sensitive zwitterionic diblock copolymer with a tertiary amine moiety, which would deprotonate upon a pH change from 2 to greater than 6. The deprotonation resulted in a loss of positive charge and the polymer block becoming water insoluble, which in turn drove the formation of vesicles due to a stronger contribution from the free energy of transfer term. The second term is the positive contribution from the interfacial free energy. This corresponds to the unfavorable partial exposure of the hydrocarbon tails to the aqueous solvent. Intuitively, it is the product of the interfacial free energy per area, σ, and the surface area per molecule of the aggregate, a. As the surface area increases, the distance between hydrophilic head groups expand, leaving more room to expose the hydrophilic tail to the solvent, resulting in an unfavorable increase in free energy. The third term accounts for the repulsive interaction between the head groups of the surfactant, due to steric and electrostatic interactions. This value is inversely proportional to the effective surface area, a. As one would expect, increasing the distance between the head groups would decrease the repulsive head group interaction contribution. Notably, the second and third terms compete to decrease and increase the effective head group area, respectively, until a balance is achieved at equilibrium.
It should also be noted that these free energy components can be expressed in a membrane lateral pressure profile. Integrals of the pressure profile function can be used to determine membrane properties such as bending rigidity, spontaneous curvature, and Gaussian curvature, which are important parameters in vesicle formation.20,21 Although there is a good amount of theoretical discussion about the translation of surfactant structure into a pressure profile,22,23 unfortunately obtaining empirical evidence has proven difficult, and at this point, the lateral pressure profile does not serve well to predict vesicle formation.
As mentioned previously, we can minimize the dimensionless free energy change
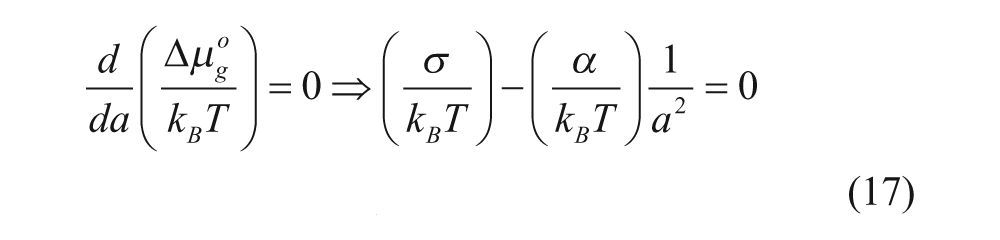
at a = a o

where a o is defined as a at equilibrium. Note that the derivative can be taken with respect to either a or N because they are related to each other. Also, as a o in equation 18 is the same as a o in the packing parameter, the effects of changing repulsive head group interactions (α) and interfacial free energy (σ) on determining the self-assembled supermolecular structure can be predicted.
In general, there are two approaches to manipulate the surface area per molecule a o : modifying the head group structure or altering the solvent conditions. For the former approach, increasing steric repulsions between head groups will increase α, thus increasing a o and decreasing the packing parameter value P, resulting in micellar aggregates. 24 Manipulation of the electrostatic interactions between surfactants can be achieved by changing the head group moiety. Adding an ionic surfactant will increase α, leading to an increased a o and a corresponding lower P value. Conversely, adding a mixture of anionic and cationic surfactants will result in a lower α (relative to a pure ionic composition) and may lead to vesicle formation, commonly known as catanionic vesicles. 25 Interestingly, an equimolar mixture of cationic and anion surfactant results in a rigid bilayer membrane, making it difficult to form vesicles. 26
The second approach to influence the surface area per molecule is to affect the surfactant head group interactions by changing the solvent. Adding salt to a solution of ionic surfactant will lead to the screening of charges, which results in reduced electrostatic interactions between like-charged head groups. 27 This will lower α and effectively increase the value of P, resulting in the aggregates tending toward vesicle morphology. The salt ions also decrease a o by disrupting the highly ordered packing of water molecules around the surfactant head groups, known as the solvation shell, which in turn reduces the intermolecular steric repulsions. Increasing the solution temperature can also decrease steric repulsions, resulting in a lower α and ultimately a higher P value. 28 In this case, the increase in temperature disrupts the solvation shell by increasing the thermal energy, which severely inhibits hydrogen bonding as hydrogen bonding is very dependent on the directionality of the chemical groups involved. Adding an organic solvent to the aqueous solution will also lower σ by reducing the energetic penalty associated with the water interaction with the exposed hydrocarbon chain. This allows for a greater a o value that leads to aggregates tending toward micellar morphology. This is why the inverse (i.e., decreasing the organic solvent to water ratio), either by adding water or by removing organic solvent, is a common vesicle formation method. 29
In the past, it was generally accepted that altering the hydrophilic head group interactions was the only practical way to control the packing parameter and, therefore, the aggregate morphology. This was in part due to the relatively constant relationship between the volume of the hydrophobic chain (v) and its critical chain length (l c ) for liposomes. Specifically, changing v generally leads to a proportional change in l c , and vice versa, for liposomes. However, this proportionality does not always exist for polymersomes as they typically have a wider variety of hydrophobic chain structures, and this leads to more flexibility in vesicle design for polymersomes.
Drug Loading
Introduction
Chemotherapeutic drugs can be encapsulated within the vesicles, where they are shielded from the harsh environment of the body. Hydrophilic drugs are encapsulated in the inner aqueous core, whereas hydrophobic drugs are encapsulated in the hydrophobic membrane region. Several methods have been developed for loading drugs into vesicles. Among these, models for loading by pH gradient, also known as remote loading, are especially prevalent. Kinetic and equilibrium models for remote loading will be discussed, as well as a model to describe encapsulation of drugs based on the fraction of the total volume associated with the aqueous cores of the vesicles.
Note that although they will not be summarized here, many models have also been developed to describe drug loading in micelles and that these models could be modified to be applicable to vesicles for hydrophobic drugs that partition into the membrane.11,30–32 Also note that Barenholz and coworkers 33 developed a flowchart for characterizing drug loading, which is beyond the scope of this review as well.
Remote Loading
Drug loading into vesicles has been achieved through pH gradients, which are sometimes accompanied by salt gradients, in a process called remote loading. Some chemotherapeutic drugs, such as doxorubicin, are neutral at neutral pH and positively charged at acidic pH. By maintaining a pH that is neutral or basic outside the vesicle, doxorubicin remains neutral and can cross the hydrophobic membrane of vesicles. By simultaneously preparing vesicles with an acidic interior, the drug becomes protonated inside the vesicles and, due to its positive charge, can no longer cross the membrane to escape the aqueous core, as shown in Figure 2 .
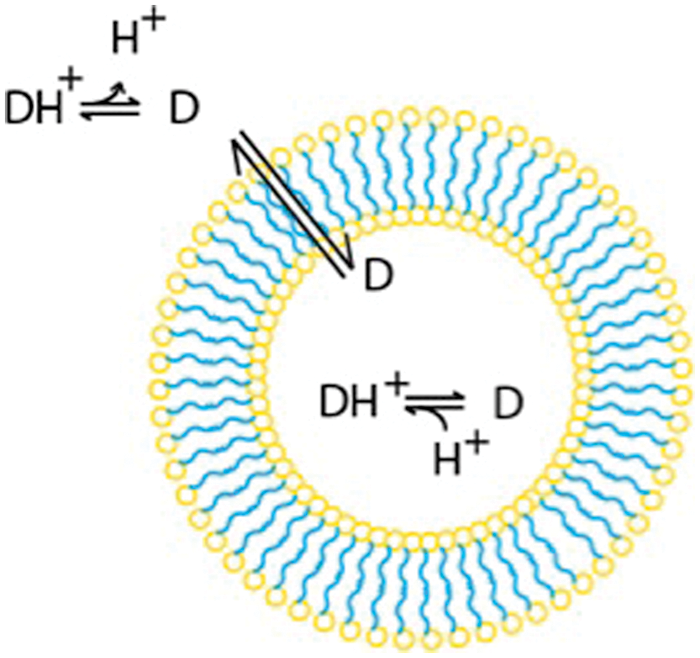
Model of remote loading. The neutral form of the drug permeates the membrane. The drug is then protonated in the acidic interior of the vesicle, and the protonated form is trapped inside the vesicle.
Several relevant models have been developed regarding the effects of various parameters on the loading efficiency and the rate of loading using the pH gradient. A model was developed by Cullis and co-workers to determine the time constant for drug loading. 34 Their model divided the vesicle into four regions: external aqueous, outer membrane, inner membrane, and internal aqueous. These regions will be denoted with the subscripts out, m-out, m-in, and in, respectively, as shown in Figure 3 . Assuming that the membrane is permeable only to the neutral form of the drug, the following expression can be written considering a mole balance on drug in the exterior solution:
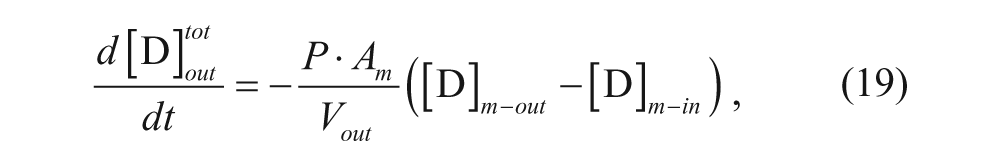
where
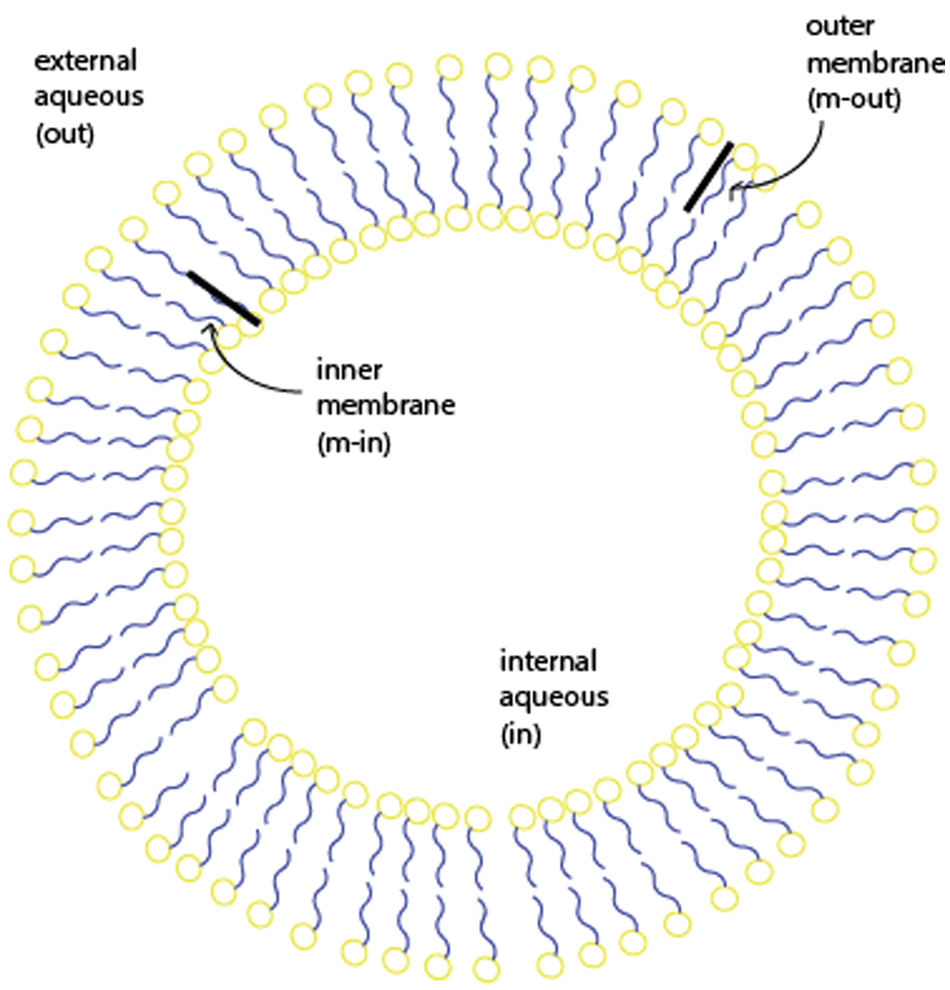
The regions of the vesicle.
The mass balance on the drug in the external and outer membrane regions can also be written as
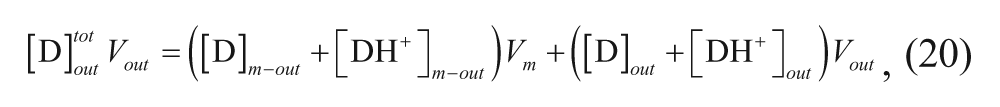
where V
out
is the volume associated with the outer portion of the membrane,
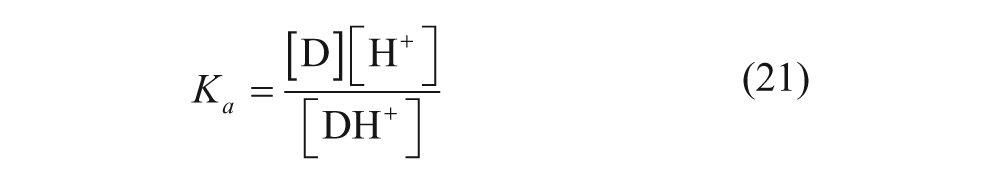
and substituting it into equation 20 yields
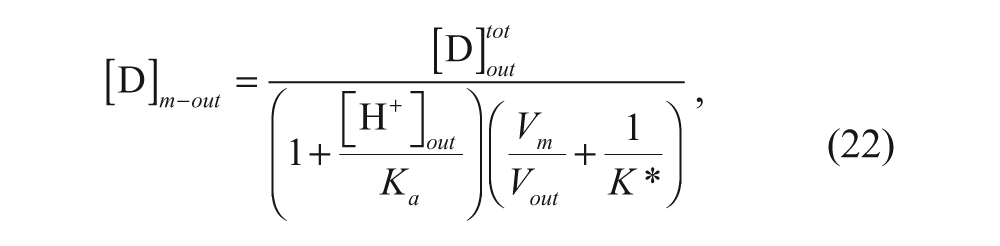
where K* is the membrane-water partition coefficient given by
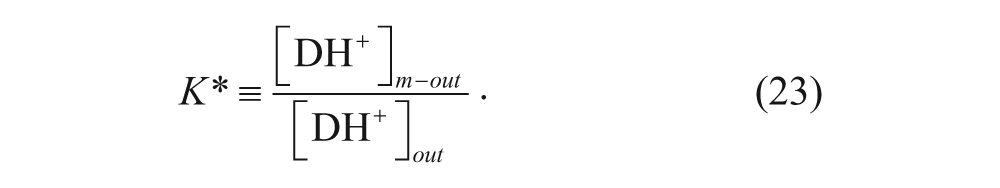
When [H+]out >> K a and V out >> V m , the expression simplifies to
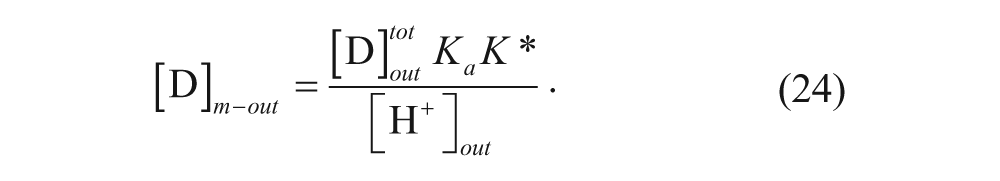
Substituting equation 24 into equation 19 and neglecting the concentration of the drug in the inner membrane portion yields
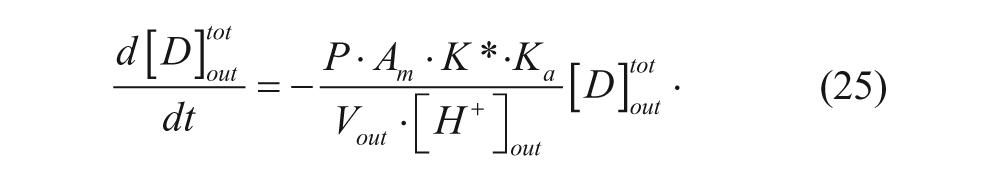
Integrating equation 25 yields
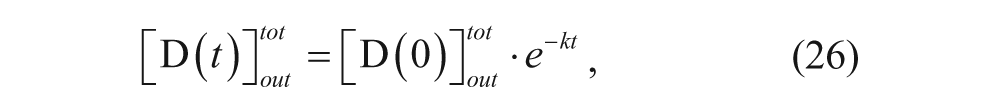
where
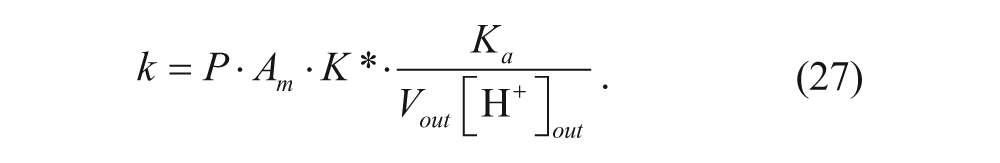
The model was confirmed for doxorubicin in large unilamellar vesicles, with a rate constant of 3·10−3s−1 at 33.4 °C. The time constant for the reaction is 1/k, which is approximately 5 to 6 min. Loading procedures should therefore run for at least 30 min to ensure maximum encapsulation.
Permeability Coefficient
To use the model presented by Cullis and coworkers, 34 the permeability coefficient is necessary. The permeability coefficient of a drug through a membrane is given by

where J is the diffusive molar flux,

where D d is the diffusivity of the drug through the membrane, K* is the partition coefficient between water and bulk organic solvent, and h is the thickness of the membrane. Note that equation 29) can be derived by assuming a linear concentration profile.
Loading Efficiency
In addition, several models have been developed to predict the loading efficiency. Two models in particular are relevant to remote loading. One thermodynamic model was developed by Hubbell and Cafiso, 36 which divided the vesicle into the same four regions discussed above. The chemical potential of the drug can be written as
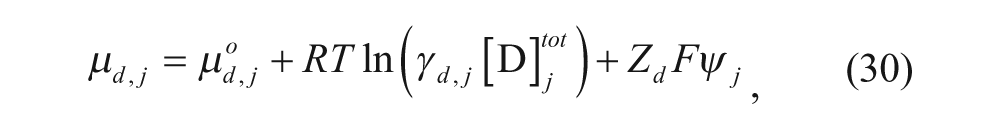
where µ
d,j
is the chemical potential of the drug in region j (where j can be any one of the four regions), µ
d,j
° is the standard state chemical potential of the drug, γ
d,j
is the activity coefficient of the drug in region j,
The ratio of concentrations in two different regions j and k (where j and k can be any two of the four regions), can be written as
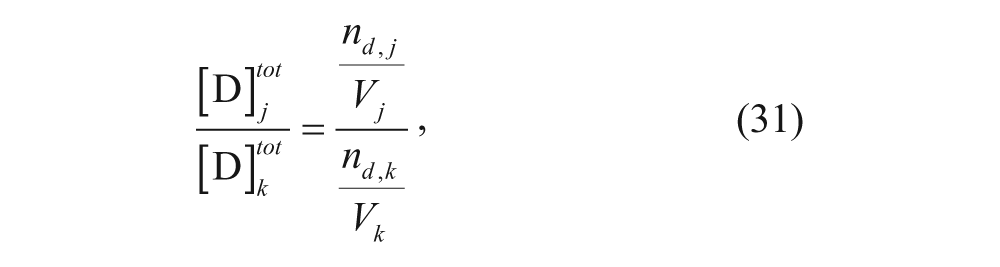
where n is the number of moles in each region, denoted by subscript, and V is the volume of each region, also denoted by subscript. At equilibrium, the chemical potentials in the regions are equal in two different regions j and k (where j and k can be any two of the four regions):
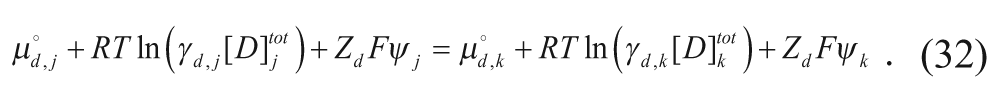
Assuming that the activity coefficients are equal for each region,
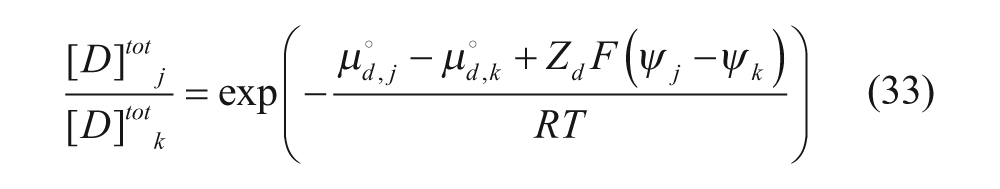
and using equation 31,
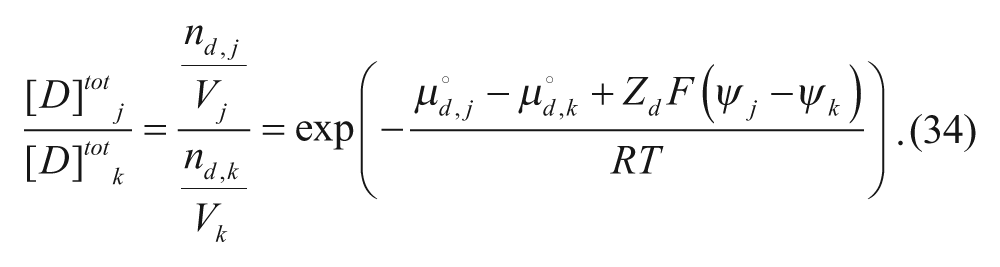
In addition, ψ out is set equal to zero as it is considered the reference potential in these problems. The electrostatic potentials in the outer and inner membranes, ψm-out and ψm-in, respectively, are then set equal to fractions of ψin.

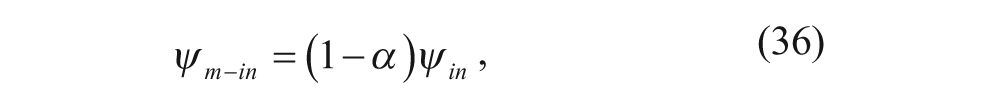
where α is a number between 0 and 1.
Using equations 34 to 36, the following expression can be derived for the ratio of moles of drug that are membrane bound (n d,bound ) to moles of drug that are free (n d,free ):
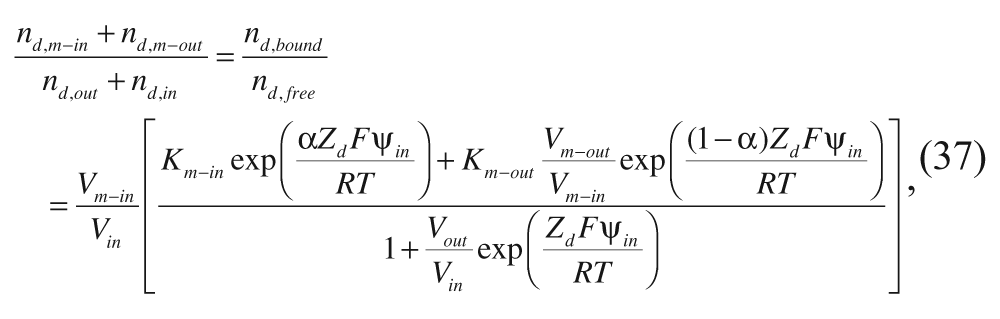
with the partition coefficients in the absence of an electrostatic potential,
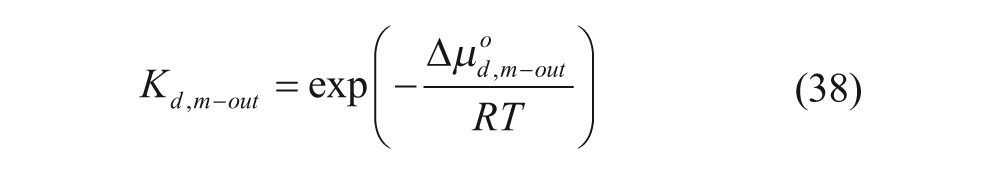
and
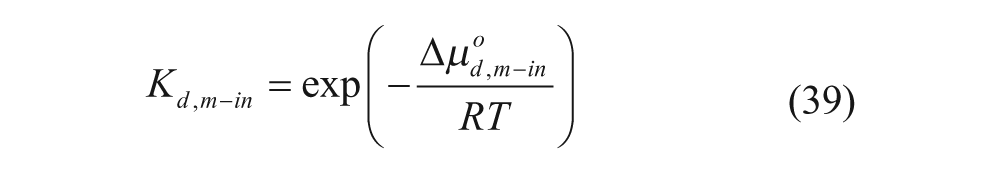
and with Δµ°d,m−out and Δµ°d,m−in representing the free energies of transfer of the drug from the aqueous phase to the corresponding membrane phase. Hubbell and Cafiso’s equation 37 does not differentiate between molecules inside the aqueous core and in the external aqueous solution because moles of drug that are free can be in both locations. However, it laid the groundwork for later models, such as that of Cullis and coworkers, 34 which will now be described.
Cullis and coworkers 34 considered the same four regions: the external aqueous region, the outer membrane, the inner membrane, and the inner aqueous core. They developed a model to predict loading efficiency, and their derivation began with the following expression for the membrane-water partition coefficient:
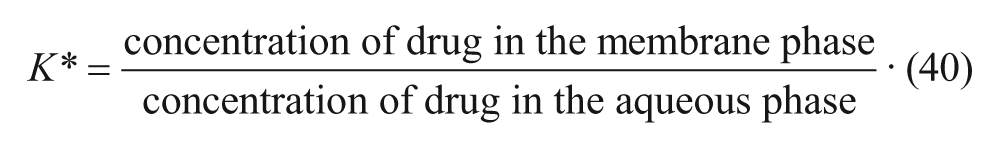
Considering partitioning of charged drug between the outer membrane and the external aqueous solution and partitioning of the charged drug between the inner membrane and the inner aqueous solution
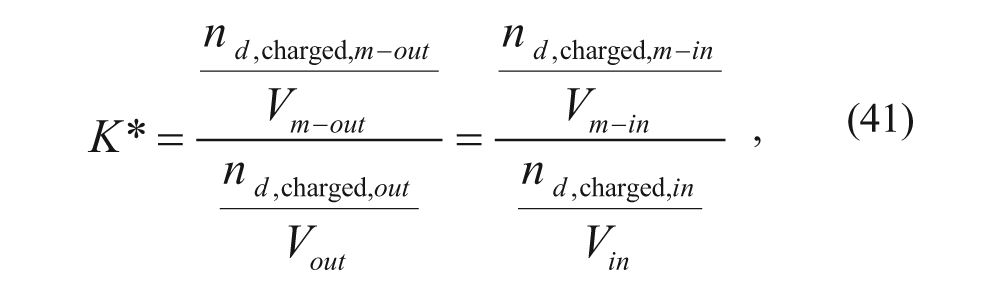
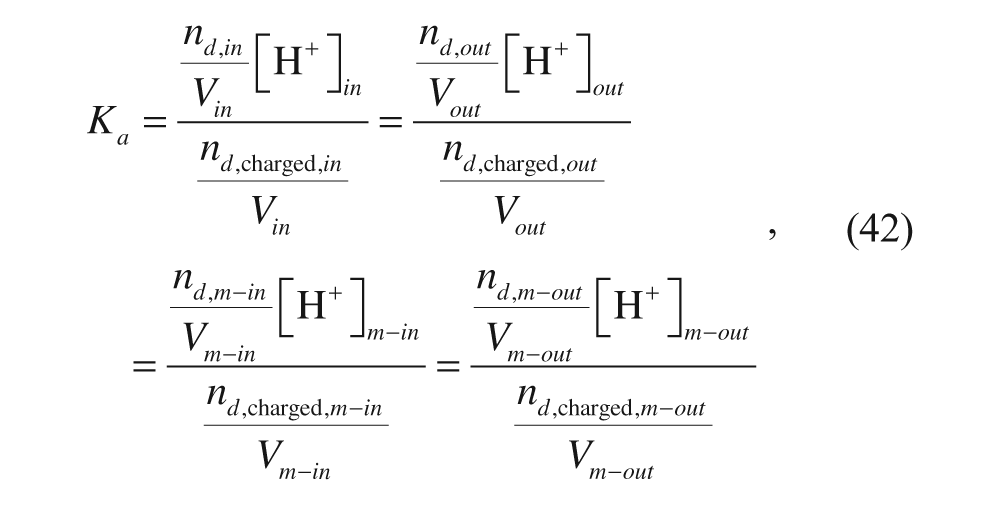
where nd,charged is the number of moles of protonated drug in each region, denoted by subscript, and V is the volume of each region, also denoted by subscript. Assuming that the acid dissociation constant for the drug is the same in all regions,
where n
d
is the number of moles of the neutral form of the drug each region, denoted by subscript, and
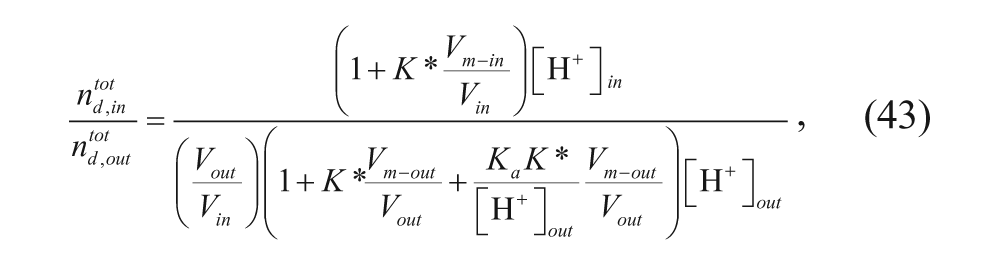
where
V
m-in
was then assumed to be equal to V
m-out
, and a new parameter K, defined as (1/2)(K*), was introduced. The lipid was then assumed to be at a low concentration, so
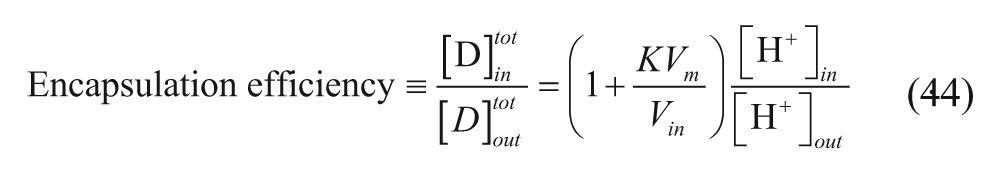
Thus, encapsulation can be maximized using a strong pH gradient because a large ratio of leads to increased encapsulation efficiency. Note that Ceh and Lasic 37 derived another model for predicting the ratio of drug encapsulated, when the drug to be loaded has multiple titratable groups.
Alternative Strategies
Alternative strategies for loading drugs into vesicles include other methods, such as thin film hydration, 38 nanoprecipitation, 39 and sonication. 40 Burgess and coworkers 38 developed a model that predicts encapsulation based on the assumption that the drug partitions evenly between the external aqueous solution and the internal aqueous core.

The model assumes that (1) all vesicles are spherical, (2) all vesicles contain a single lipid bilayer, (3) the drug does not interact with the lipid at the interface, and (4) the particle size follows a log-normal distribution.
For a vesicle with radius r i and membrane thickness h, the outer and inner surface areas are given by

and
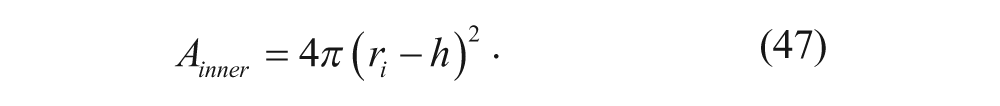
Using the average lipid molecular area (a), the number of lipid molecules per vesicle is given by
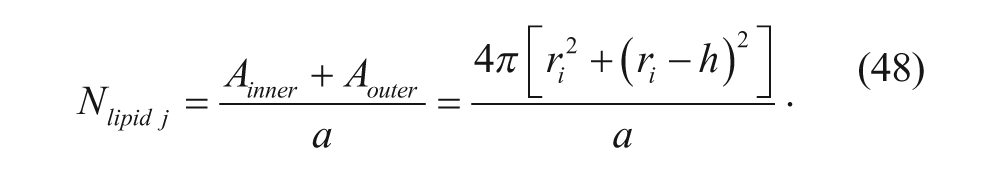
If the mean particle size, µ, and size distribution, σ, are known, the probability, P i , that a vesicle has size r i is equal to
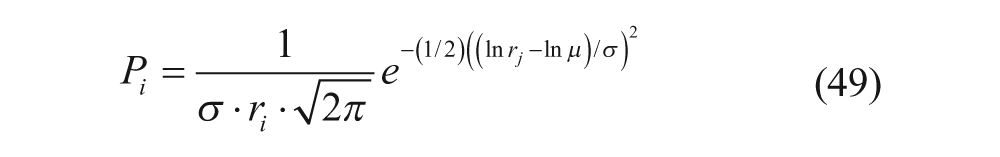
for a log-normal distribution. Thus, the total number of vesicles can be calculated using the concentration of lipid, c, and the total volume, V tot .
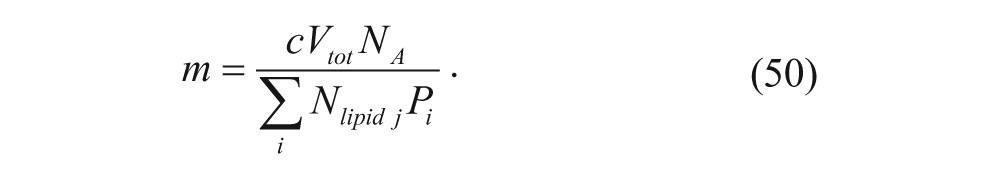
The number of vesicles of a given size is given by

The total internal volume, V in , is then given by

where v i is the internal volume of a vesicle, given by:
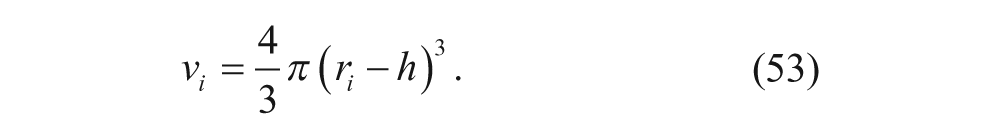
Finally, an expression for the encapsulation efficiency, given by equation 45, can be derived by combining equations 48 and 50 to 53.
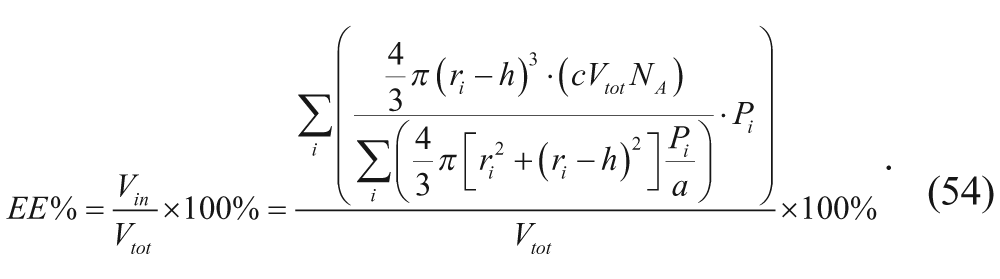
Equation 54 dictates that maximum encapsulation efficiency is achieved at high vesicle concentrations (large c).
Drug Release
Introduction
In analysis of the release from vesicles, math models are useful for investigating release kinetics. Models for release kinetics are available for leakage and passive diffusion through the membrane.41,42 In addition, vesicles can be designed to degrade at the tumor site because of its particular pH environment.19,43 A model analyzing pH-sensitive vesicles is also presented. 43
Diffusion
Higuchi 41 performed early modeling work in drug release from polymers. The Higuchi model characterizes drug release as
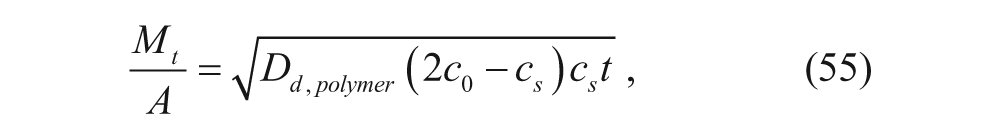
where M t is the total moles of drug released at time t, A is the total surface area of the polymer, D d,polymer is the diffusivity of the drug in the polymer, c0 is the initial concentration of drug in the polymer, and c s is the solubility of the drug in the polymer. This model assumes the following: (1) The initial concentration of drug must be much higher than the solubility of the drug in the polymer. (2) Diffusion is one dimensional. (3) The drug is much smaller than the thickness of the membrane. (4) There is negligible modification to the polymer (e.g., swelling or degradation). (5) The diffusivity is constant. (6) Perfect sink conditions are maintained. For drug delivery, diffusion out from the vesicle is undesirable until the vesicle reaches its target. Therefore, to minimize unwanted drug release according to equation 55, the diffusivity of the drug in the membrane should be minimized.
Ritger and Peppas 42 developed a more accurate model, commonly called the power law model. This model added a first-order term to the t1/2 dependence previously found in the Higuchi model:
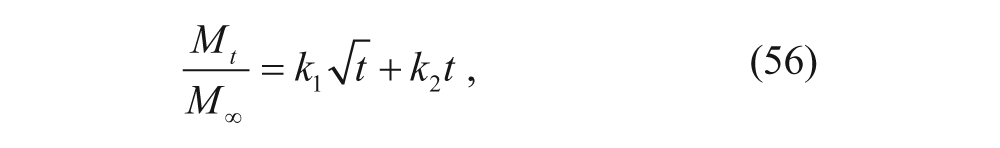
where M t /M∞ is the percentage of drug released at time t, and k1 and k2 are rate constants. Multiple geometries were considered, and the following expression was obtained:

where n is called the diffusional exponent, which varies based on the geometry, and k 3 is a rate constant.
Another approach to estimate this rate of drug release involves solving the conservation of species equation in spherical coordinates with no convection and no homogeneous reaction, which is given by
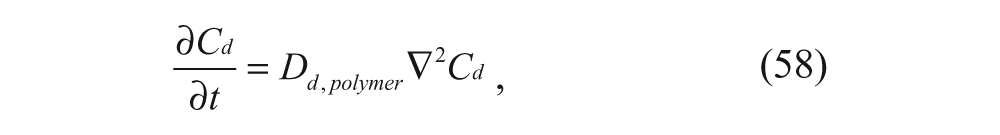
where C d is the concentration of the drug as a function of time and radial position, D d,polymer is the diffusivity of the drug in the polymer, and r is the radial distance from the center of the vesicle. The initial condition is given by

for 0 < r < R, where C1 is the initial concentration and R is the radius of the vesicle. Two boundary conditions are also required. The first boundary condition states that the flux through the center of the vesicle is zero. The second boundary condition is given as
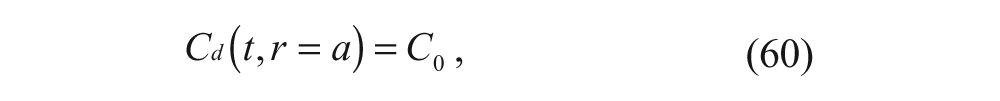
where C0 is the concentration in the membrane.
This model is not entirely descriptive of vesicles, as it does not account for the inner aqueous core. To adjust for the inner aqueous compartment, a second conservation of species equation should be used for the aqueous core.
Release by Vesicle Degradation
One method to maximize drug delivery, without unwanted leakage from vesicles, is through degradable vesicles. This can be achieved through pH-sensitive,44,45 ultrasound-sensitive, 46 oxidation-responsive, 47 hydroxypropyl methylcellulose swellable vesicles, 48 and temperature-sensitive 49 vesicles. This review will cover only pH-sensitive vesicles.
The ideal pH-sensitive vesicle for intracellular delivery will release its contents rapidly at a pH between 5 and 6 as seen in the endosome while retaining its contents at the neutral pH 7.4 of the blood. Note that tumors also have a lower extracellular pH (<6.5) than healthy tissue. 50 One strategy to achieve this release behavior involves using lipids with acidic amphiphiles that act as stabilizers at neutral pH but are degraded at acidic pH. 43 Another approach uses a caged headgroup that changes size at acidic pH, destabilizing the vesicle. A third strategy uses titratable polymers to trigger membrane destabilization. 51
Szoka and coworkers 44 developed a pH-responsive vesicle using PEG-orthoester-distearoylglycerol (POD). At neutral pH, the liposome is stable due to steric repulsions between liposomes due to the PEG. At acidic pH, such as that found in endosomes, the POD is hydrolyzed to release PEG, resulting in aggregation and leakage. The aggregation of vesicles leads to fusion, which alters vesicle shape and therefore makes the vesicles more porous. The release kinetics were characterized by a slow release phase, termed the lag phase, followed by a rapid burst phase, as shown in Figure 4 . 52 The key parameter in this model is the duration of the lag phase because it is desirable for this time to be larger than the time it takes to reach the tumor for vesicles targeting cancer.
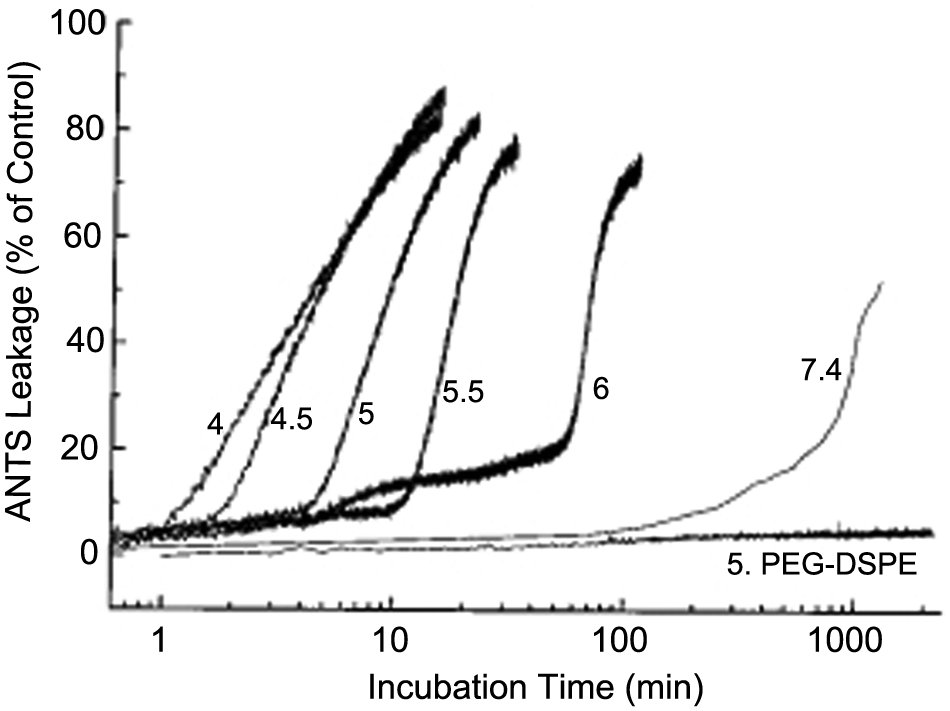
Drug leakage profiles from pH-sensitive liposomes in various buffers. The release is slow during the lag phase and followed by rapid release in the burst phase. Reprinted with permission from Bioconjugate Chemical Society, 12, Guo, X., Szoka Jr., F. C. Steric stabilization of fusogenic liposomes by a low-pH sensitive PEG-diortho ester-lipid conjugate, 291-300, 2001. Copyright 2001 American Chemical Society.
Assuming constant pH in the external aqueous solution, 52
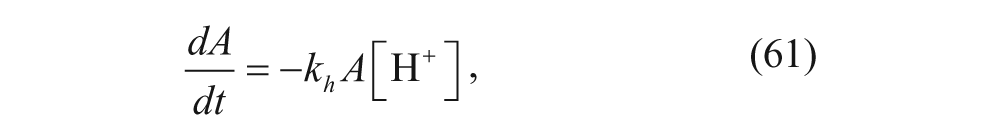
where A is the number of stabilizing hydrolyzable groups (POD) on the surface, k h is the rate constant for ortho ester hydrolysis, and [H+] is the concentration of protons in the aqueous solution. Integrating equation 61 yields
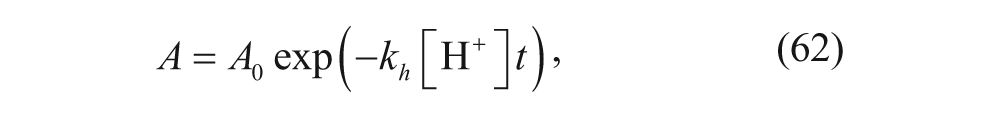
where A0 is the initial number of POD. Experimentally, Szoka and coworkers found that the rapid release phase begins once the variable A reaches a critical number, A c . Therefore, the duration of the lag phase, t1, is predicted to be
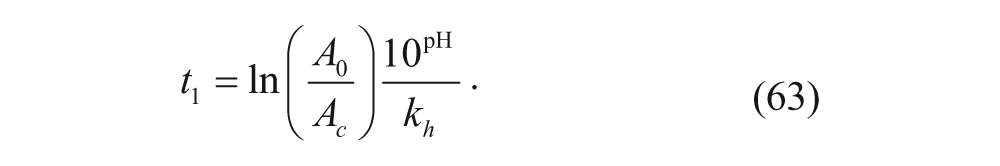
Accordingly, equation 63 suggests that increasing the number of stabilizing hydrolyzable groups increases the lag phase, further ensuring minimal leaking until the tumor is reached.
Conclusion
This article reviewed mathematical models that have been derived for further understanding vesicle formation and stability along with predicting drug loading and release. With regard to vesicle formation and stability, the molecular packing parameter is extremely important. Not only does it enable a higher degree of control during the formation process, it also helps predict changes in morphology during delivery, which is crucial in a field where the vesicles’ environment is dictated by physiological conditions, such as pH, oxidative stress, and temperature. To avoid potential problems, many techniques have been developed to increase or maintain stability during delivery. For example, PEG has been conjugated to vesicles to increase circulation time. 53 PEG protects the vesicle from uptake by the mononuclear phagocytic system (generally referred to as having “stealth” properties) and by providing steric stabilization to the vesicle structure. It also has been shown that the presence of additives that intercalate the vesicle wall, such as cholesterol and cardiolipin, will increase the stability of liposomes. 54 Vesicles can also be stabilized after formation by cross-linking the surfactant molecules with each other. For example, Meier and coworkers 55 used ultraviolet irradiation to increase vesicle stability by intravesicular cross-linking of the polymerizable ends of triblock copolymers. Similarly, increasing the molecular weight of the amphiphile will increase stability due to enhancing van der Waals attractive interactions between the chains, particularly for polymersomes. This alteration also results in decreased permeability and fluidity of the vesicle membrane. 10
Although maintaining vesicle stability during drug transit is important, facilitating vesicle destabilization at the target site is useful for efficient delivery. To this end, thermodynamic considerations can be used to establish important design criteria. For example, a target site may have a significantly higher or lower pH than the transit environment. To use this potential transition, Deming and coworkers 56 created a block copolypeptide vesicle designed so that the hydrophobic segments had protonatable lysine groups with a pKa in between the pH transition values. When the vesicles encountered a lower pH, the hydrophobic core became protonated, which led to destabilization of the vesicles through the positively charged electrostatic repulsions and the increased solubility of the polypeptide chains.
With regard to drug loading, the kinetics and equilibrium of drug loading can be derived in terms of the partition coefficient, the diffusivity of the drug in the membrane, the thickness of the membrane, the pKa of the drug, the pH inside and outside the vesicle, and the time of drug incubation. With this quantitative understanding of drug loading in vesicles, researchers will be able to engineer the vesicles and use an appropriate drug incubation time to achieve a target drug loading value. With regard to release from the vesicles, it can be viewed as either simple diffusion, corresponding to leakage, or rapid release, corresponding to stimuli-responsive vesicles that can be triggered by changes in the environment. For simple diffusion, models from the Higuchi and Peppas groups have been used to correlate the relationship between vesicle parameters and observed leakage rates. For pH-responsive vesicles, Szoka and coworkers developed a model that considered both the kinetics of a slow lag phase as well as a rapid burst phase and, in particular, quantitatively derived a relationship that demonstrated the importance of the number of hydrolysable groups. All of these drug release models can be used to optimize and engineer vesicles for minimal leakage during transit and maximum release at the target site.
Mathematical models can be used to predict the effects of varying vesicle properties on the formation, stability, and ability to encapsulate and release drugs. Such quantitative relationships can help save time and resources in the development of vesicle-based drug carriers. It is our hope that researchers can use the models in this review to complement their existing experimental tools to generate more efficacious drug delivery systems.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors would like to thank the support of the Department of Defense Prostate Cancer Research Program under award number W81XWH-09-1-0584 and the National Science Foundation DMR 0907453.