
Abstract
With an aim to develop peptide-based protein capture agents that can replace antibodies for in vitro diagnosis, an ultra-high-throughput screening strategy has been investigated by automating labor-intensive, time-consuming processes that are the construction of peptide libraries, sorting of positive beads, and peptide sequencing through analysis of tandem mass spectrometry data. Although instruments for automation, such as peptide synthesizers and automatic bead sorters, have been used in some groups, the overall process has not been well optimized to minimize time, cost, and efforts, as well as to maximize product quality and performance. Herein we suggest and explore several solutions to the existing problems with the automation of the key processes. The overall process optimization has been done successfully in orchestration with the technologies such as rapid cleavage of peptides from beads and semiautomatic peptide sequencing that we have developed previously. This optimization allowed one-round screening, from peptide library construction to peptide sequencing, to be completed within 4 to 5 days. We also successfully identified a 6-mer ligand for carcinoembryonic antigen–cell adhesion molecule 5 (CEACAM 5) through three-round screenings, including one-round screening of a focused library.
Introduction
It is of utmost significance and utility to equip reliable agents that bind strongly and specifically to target biomarkers for both research and clinical applications. Despite widespread utility of antibodies associated with their compelling advantages, including high fidelity, various limitations prevent researchers and clinicians from employing them for universal tools as protein capture agents for diagnosis.1,2 Basically, conventional antibodies are laborious and costly to produce under a skill-demanding biological platform, which often turns out not fruitful despite huge resources invested. Furthermore, antibodies, as part of protein families, must retain a precise folding structure to function properly, limiting the conditions under which they can be stored and employed. Thus, it is a broad consensus that there is a high demand for other classes of molecular agents of high affinity and high specificity that can potentially replace antibodies.
Nonantibody protein capture agents have been investigated in recent years. The typical types of such alternative agents are nucleic acids aptamers (DNAs and RNAs), 3 natural peptide libraries by phase display technique, 4 and small molecules that are often related to drug discovery and development, whereas other classes of molecular families such as lipids, carbohydrates, inorganic materials, and even other proteins also can be included. Nucleic acid aptamers hold possibility with the early stage promises but possess the intrinsic problem of limited chemical diversity, as there are only four standard bases as compared to more than 100 amino acid entities in synthetic peptide libraries. Other issues, such as upscale production and physiological stability, comprise additional hurdles in the universal applicability of nucleic acid aptamers. On the other hand, peptides selected from phage display libraries can offer reasonably good performance in conjunction with a high degree of diversity they can provide. However, the naturally occurring L-amino acids comprising such peptides are not stable enough for proteolytic cleavage under physiological conditions. It is not feasible to incorporate nonnatural or artificial amino acids, as the microorganism-based library generation involves a biological platform, which may limit facile production at the stage of downstream applications. Finally, small-molecule ligands can exhibit a high affinity for their protein targets. However, achieving good affinity and selectivity remains extremely difficult, thus compromising their utility for detection purposes.
A promising alternative is synthetic peptide-based affinity agents that are identified using one-bead-one-compound (OBOC) libraries.5,6 The advantages of OBOC peptide libraries include well-established synthesis, high diversity, chemical stability, and easy modification by using nonnatural or artificial amino acids due to the chemistry platform. 7 This diverse and flexible platform can offer good biochemical, chemical, and physical stability and water solubility. However, typical OBOC libraries, bearing 4 to 10 peptide bonds, can express only 10 5 –10 8 diversity and require a lot of time and labor to obtain peptides with acceptable binding affinities. 8 The number of peptides that can be identified through a manual process or peptide sequencing by Edman degradation9,10 is quite limited. Clearly, there is an urgent need for a real high-throughput screening (HTS) process. Herein, we demonstrate an ultra-HTS platform through automation and optimization, including rapid and robust peptide sequencing by using matrix-assisted laser desorption ionization tandem time-of-flight (MALDI-TOF/TOF).
Results and Discussion
In the development of an ultra-HTS technology for OBOC combinatorial peptide libraries, one of the most important factors is to decide how to sort positive peptides in a rapid and reliable fashion from an assay, which is often the most time-consuming process. One potential method is to use an optical detection method to quickly identify and sort positive beads with strong fluorescent intensity through interactions between dye-labeled proteins and peptides on beads. In addition to the optical detection method by using a dye-label target protein, we can adopt a cyanogen bromide (CNBr)–cleavage method to cleave peptides from a bead for structure analysis due to its simplicity and efficiency. Compared with other approaches such as ladder synthesis11–14 and Edman degradation/mass spectrometry (MS) sequencing,15–19 the CNBr-cleavage method in conjugation with de novo sequencing can be simpler and faster. In relation, we reported on a rapid CNBr-cleavage method under microwave irradiation to cleave most peptides from a bead within 1 min. 20 Therefore, an ultra-HTS platform can be established by the combination of automatic instruments for fluorescence-based screening and de novo peptide sequencing via CNBr-cleavage ( Fig. 1 ). Herein, we discuss several major issues with automation and suggest solutions to problems found from screening against various types of proteins. In particular, we focus on three major steps: the construction of peptide libraries, automatic bead sorting, and peptide sequencing. These major steps have been automated with the commercial instruments and optimized by tackling key issues to make each process more efficient in time and cost. To make the three key steps automatic, we selected Titan 356 from AAPPTec (Louisville, KY) for automatic peptide library construction, COPAS Plus from Union Biometrica (Holliston, MA) for automatic bead sorting, and ultrafleXtreme MALDI-TOF/TOF from Bruker (Billerica, MA) for automatic MS and tandem mass spectrometry (MS/MS) data acquisition.
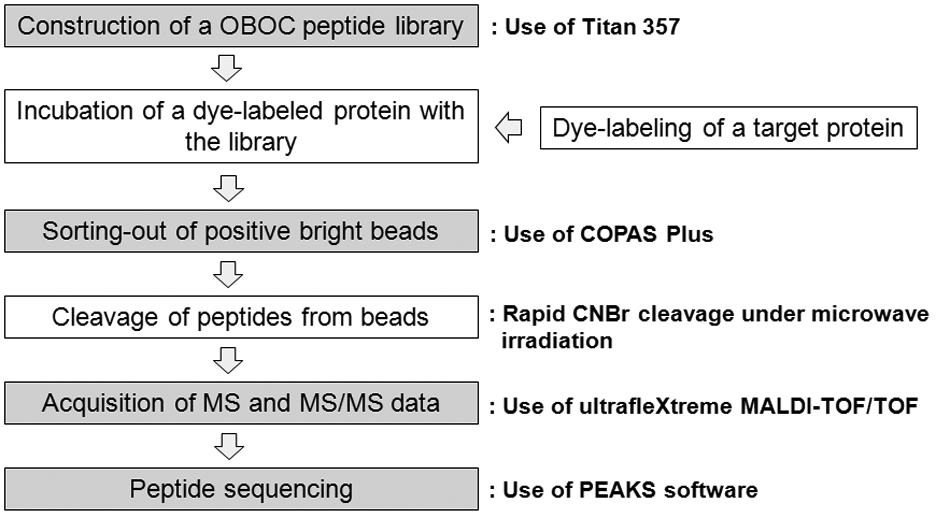
Overall screening process of a one-bead-one-compound (OBOC) peptide library. The shaded steps were selected for automation with commercial instruments and software. CNBr, cyanogen bromide; MS, mass spectrometry; MS/MS, tandem mass spectrometry; MALDI-TOF/TOF, matrix-assisted laser desorption ionization tandem time-of-flight.
Titan 357 for Semiautomatic Construction of Bead-Based Peptide Libraries
An OBOC peptide library was constructed for screening against a target protein in a high-throughput fashion. Important considerations in the construction of an OBOC peptide library are (1) the type of beads, (2) efficient split-and-mix process, and (3) minimization of solvent and chemical consumption.
TentaGel S amino resin (RappPolymere, Tübingen, Germany) was selected as a solid support because of its wide compatibility in both organic and aqueous media, as well as its spherical and monodisperse shape (90 µm). It is suitable for both peptide synthesis in organic media and biochemical assay in aqueous media. One of the most suitable automatic synthesizers for the OBOC library generation is Titan 357 from AAPPTec. Along with the typical automatic feature to support the Fmoc-chemistry21–24–based peptide synthesis, Titan 357 is equipped with an automatic split-and-mix function associated with a second arm with a specially silanized glass probe that can handle the polymeric beads without electrostatic problems. It is highly convenient to run the equipment in constructing 5- to 7-mer libraries on a reasonable scale (~0.2–2 g). However, a fully automatic operation of Titan 357 will lead to consumption of a large amount of solvents and time to complete the split-and-mix process, without leaving the beads in the collective vessel (CV) or in the reaction vessels (RVs). The beads left over in CV will not participate in the next coupling step, so the library will contain some shorter peptides when it is fully elaborated. In splitting ~2 g of TentaGel beads from CV to 18 RVs, Titan had to perform more than seven times the transfer of beads using 50 mL of solvents comprising N-methylpyrrolidone (NMP)/dichloromethane (DCM) (50/50), which make an even suspension throughout the mixture. The time elapsed may easily amount to several hours. To circumvent these problems, we implemented manual input so that the mixing and splitting could be carried out efficiently and economically.
Mixing could be done relatively quickly and less thoroughly because a trace amount of beads left in RVs will still participate in the coming coupling reactions even if the diversity generation might be slightly compromised. In the meantime, the split should be done more thoroughly because the beads left in CV will end up being shorter than the fully elaborated peptides. Using NMP/DCM (50/50) gives a reasonably even suspension of the TentaGel beads so that any fraction of a given volume could contain a similar amount of beads. It is effective both in split-and-mix. In mixing 100 mg resin from 18 RVs to CV, for example, dispensing 2 mL of the solvents was sufficient for initial transfer, whereas two more transfers could have been done using less volume of solvents (e.g., 1 mL and 0.5 mL, successively).
Although no significant amount of beads resided in RVs, one more mixing was generally performed using 0.5 mL of DCM to ensure the complete ferrying of beads from RVs to CV. It took less than 10 min by using manual dispensing and transferring, whereas a fully automatic method needed at least 1 h for complete mixing. This difference became more conspicuous in performing the split. A fully automatic operation took more than 2 to 3 h in splitting 2 g TentaGel beads evenly into 18 RVs. The volume of solvents used to provide even and fluidic suspension was initially 50 mL, and subsequently, decreased volumes of the mixture solvent were used six to eight times. Manually, merely 15 min was required for a complete split using a minimum amount of solvents three times. Starting with 50 mL of mixture solvents at first, another two 10-mL portions of solvents were sufficient to completely transfer the beads. To ensure that no bead remained in the CV in the end, one quick wash using 5 to 10 mL DCM was performed while Titan worked for the next operation.
One drawback of running a manual split-and-mix is that it cannot be run continuously. However, significant time savings and solvent reduction could be achieved. The construction of a 5-mer and 6-mer library could be finished within an overall maximum time of 24 h and 48 h, respectively. This semiautomatic run was also favored for the good quality of peptides on beads. Methionine was introduced as a cleavable linker to adopt CNBr-mediated cleavage chemistry for sequencing based on MALDI-MS/MS. To elaborate the diversity region, 5-mer or 6-mer peptides seemed suitable for efficient synthesis and sequencing with a great number of diverse sequences (18 5 = ~1.9 million) readily generated by the “split-and-mix” approach. In general, 18 unnatural (i.e., D-) amino acids were employed as the diversity elements, excluding cysteine and methionine. It was noted that the coupling yields were variable upon the sequences. For example, the coupling reactions of hydrophobic and bulky residues proceeded slower than those of small and hydrophilic residues. Some short oligomers turned out particularly inefficient toward coupling with incoming Fmoc–amino acids. Therefore, all the coupling reactions were performed twice (i.e., double coupling). Followed by a typical removal of the Fmoc group by piperidine in NMP (20%, v/v), the cycle of mix, split, coupling, and Fmoc deprotection was repeated until the beads appended the desired length of peptides. The free amino group at the N-terminus was capped with the acetyl group (Ac) to minimize nonspecific interactions on screening as well as to increase the on-shelf stability. Finally, the protective groups in all residues were completely removed by a trifluoroacetic acid (TFA) cleavage cocktail to render the intact peptide species available for the following biochemical screening against a target protein. To confirm the purity of peptides on beads before use of a newly generated library for screening, more than 20 beads were randomly picked up, and peptides of each bead were cleaved to obtain MS and MS/MS data to investigate if there were any truncated peptides from analysis of each MS and MS/MS spectrum.
COPAS Plus for Automatic Sorting of Positive Beads
A COPAS bead sorter was used for sorting positive beads after incubation of peptide library beads with a fluorescence dye-labeled protein.14,21–24 We found that there were several important issues in sorting with the COPAS system, such as (1) the selection of a suitable fluorescence dye, (2) the degree of dye labeling, (3) the strategy of sorting, (4) the background noise level from the interaction between dye and peptide on beads, and (5) the inclusion of false-positive beads during sorting.
Red fluorescent Alexa Fluor 647 (Molecular Probes, Eugene, OR) was chosen to label a target protein because of its great photostability and resistance to photobleaching after labeling proteins.25,26 The dye, excited by a red laser, was chosen in particular because of its relatively low autofluorescence from TantaGel beads compared with dyes excited by a green (λ = 488 nm) or a yellow laser (λ = 561 nm) ( Fig. 2 ). It is noteworthy that TentaGel beads showed very strong autofluorescence from its polystyrene core on excitation with the yellow solid-state laser ( Fig. 2b ). The degree of labeling (i.e., average number of dye per protein) could be adjusted to be statistically close to 1 by controlling the labeling conditions to minimize the change of protein conformation and nonspecific interactions between dyes on a protein and peptides on a bead due to overlabeling. The degree of dye labeling could be measured by ultrafleXtreme MALDI-TOF/TOF. Figure 3 shows MS spectra of several different proteins labeled with different amounts of Alexa Fluor 647. The degree of labeling depended on the type of proteins, and overlabeling problems could be avoided by using this method. Usually, the degree of labeling fell in the range of 0.6 to 1.0 by using a one-third portion of the Alexa Fluor 647 labeling kit from the manufacturer’s protocol to label 1 mg of a target protein. Instead of a conventional diode red laser (0.5–1.0 mW), a powerful solid-state laser (640 nm, 10–20 mW) was used to compromise the decrease in fluorescence intensity due to a lower dye-labeling level.
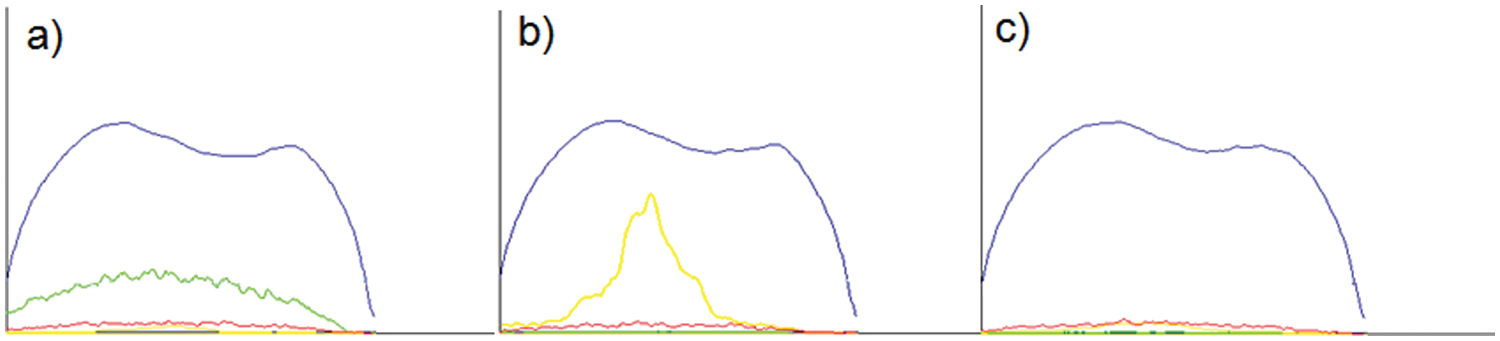
Fluroscence profile measurement of TentaGel beads with excitation source of (a) green (l = 488 nm), (b) yellow (l = 561 nm), and (c) red (l = 640 nm) solid-state laser. Relative intensity of the profiles in the graphs are EXT (blue), green (green), yellow (yellow), and red (red) fluorescence along with the length of a bead (x-axis) of each measurement.
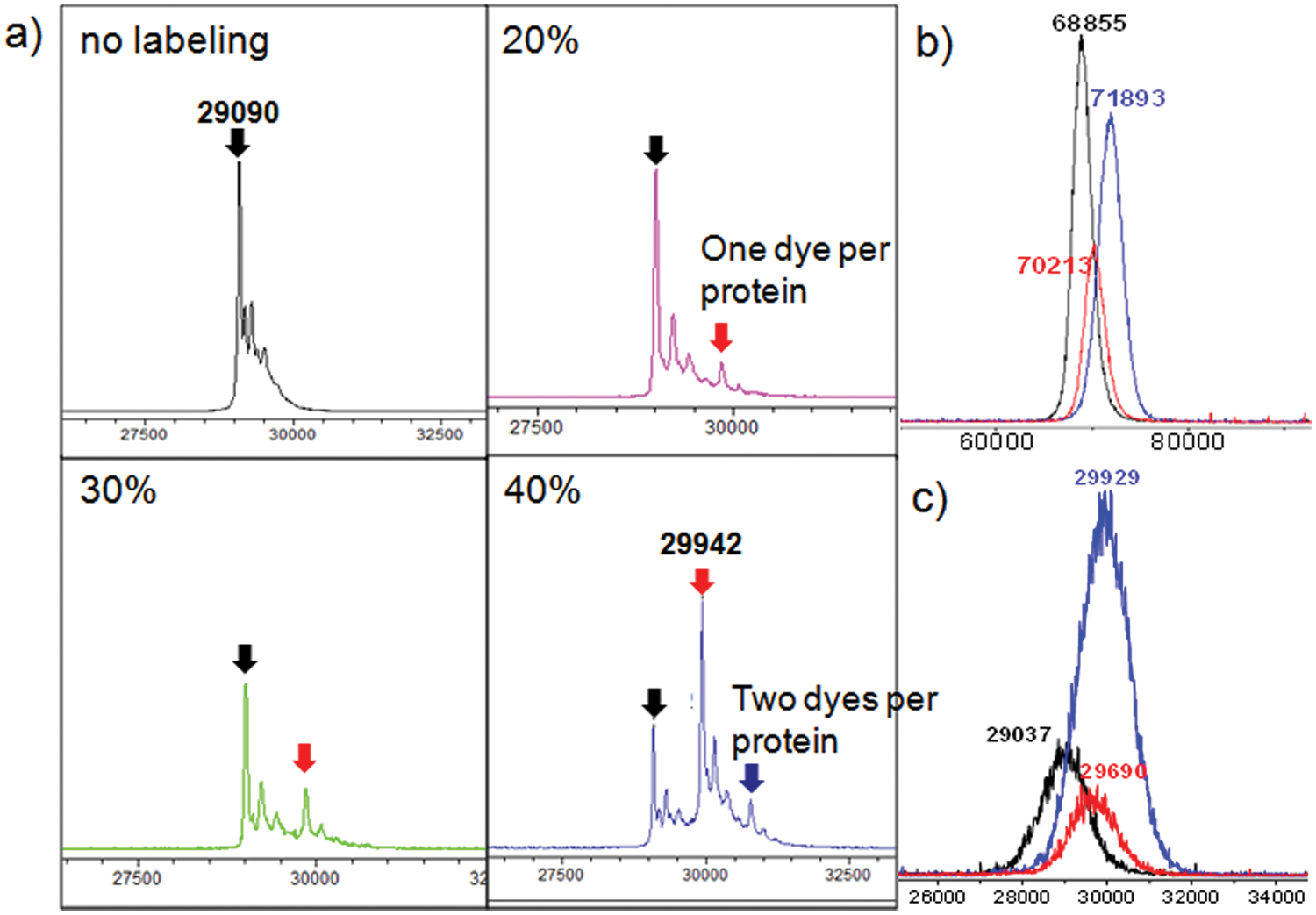
Mass spectrometry (MS) spectra of proteins labeled with different amounts of Alexa Fluor 647 by ultrafleXtreme matrix-assisted laser desorption ionization tandem time-of-flight (MALDI-TOF/TOF). (a) Bovine carbonic anhydrase (bCAII) labeled with different amounts of Alexa Fluor 647 (20%, 30%, and 40% of the recommended amount in the manufacturer’s protocol), (b) a-fetoprotein (AFP) labeled with different amounts of Alexa Fluor 647, and (c) prostate-specific antigen (PSA) labeled with different amounts of Alexa Fluor 647 (black line, no labeling; blue line, recommended amount in the manufacturer’s protocol; red line, one-third of the recommended amount).
For stringent screening, we performed a two-stage sorting protocol by modifying the previous method.21,22 The first sorting, governed in enrich mode, was carried out at a high flow rate (>80 beads/s) to collect approximately the 1500 to 2500 most fluorescent beads ( Fig. 4 ). Selection criteria such as size range (TOF), level of fluorescence intensity, and uniformity of fluorescence (RedPH, red fluorescence peak height in Profiler [Union Biometrica, Holliston, MA]) were defined by drawing polygons in the gating and sorting windows. The gating polygon was defined to exclude small-size bubbles as well as negative beads, whereas the sorting gate was defined to exclude spiked fluorescence due to electronics and detector instability. Within 2 h, uniformly fluorescent beads were collected in a dish as a consequence. However, the collected beads that mostly consisted of positive beads usually contained a significant number of negative beads because of the high flow rate process. The collected beads, which in general constituted <1% of the screened library, were washed thoroughly by deionized water to remove salts to do MALDI samplings directly after sorting of positive beads into 96-well plates and then CNBr-mediated cleavage of peptides from beads due to bad effects of salts on the acquisition of MS spectra. The second sorting, governed in pure mode, was carried out at a lower flow rate (5–10 beads/s) for 30 min. With the two-stage sorting protocol completed within 3 h, each of the highest fluorescent beads was isolated directly into 96-well plates to facilitate the following step: CNBr-mediated cleavage under microwave irradiation.
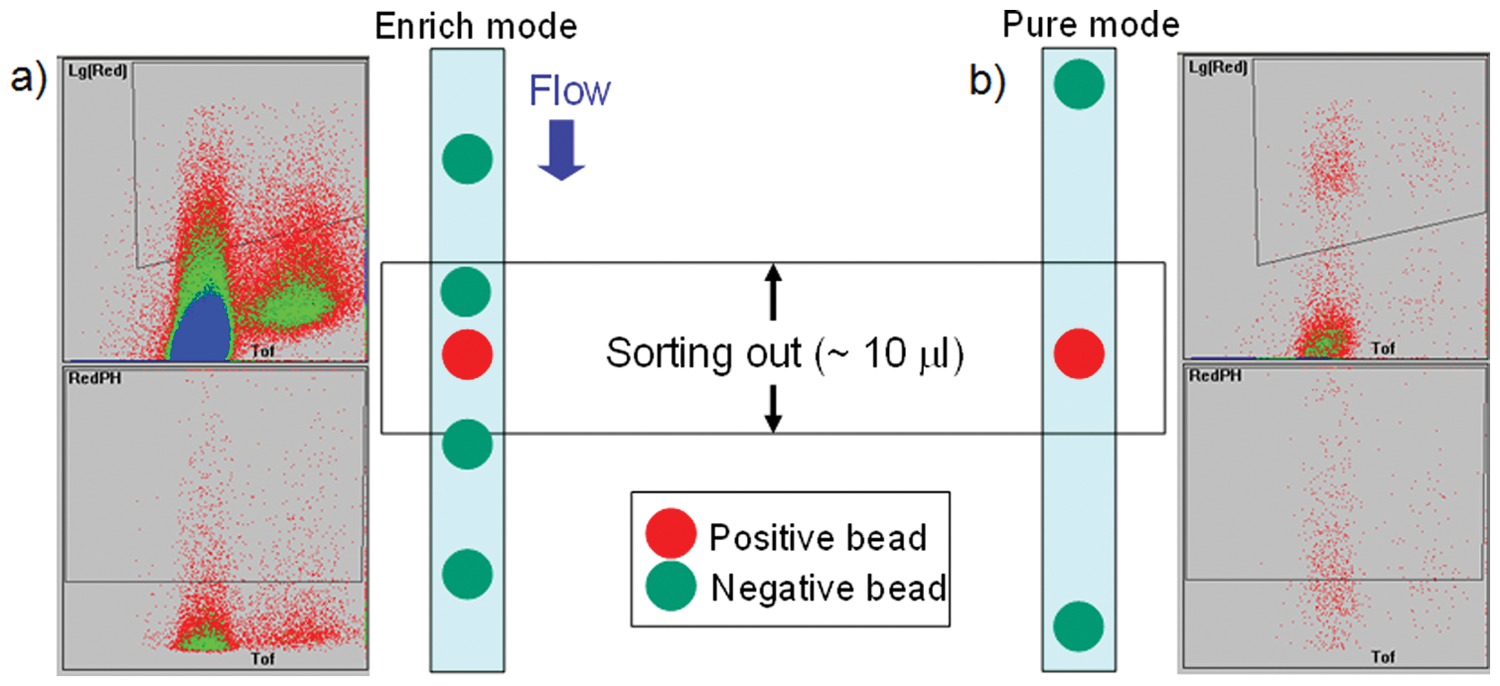
Typical sorting experiments of (a) first sorting at a high flow rate (>80 beads/s) and (b) second sorting at a low flow rate (5–10 beads/s). Upper diagrams (red fluorescence in logarithmic scale vs time of flight) show gating of beads in which the polygons define selection of gated beads by excluding bubbles and baseline fluorescent beads. Lower diagrams (red fluorescence peak height vs time of flight) show sorting of beads in which the polygons define the selection of uniformly fluorescent beads. Dot colors represent the population of the bead on each position, such as red on 1 to 10 beads, green on 10 to 100 beads, and blue on 100 to 1000 beads.
In contrast with the autofluorescence issue, other issues of COPAS sorting are related to inclusion of false-positive beads directly and indirectly. The false-positive beads can be generated by the interaction of fluorescent dyes with peptides and nonspecific interactions between peptides on beads and target proteins.23,27
The inclusion of false-positive beads due to nonspecific interactions between peptides and target proteins is not the only problem with COPAS sorting. Usually, a competing protein such as bovine serum albumin (BSA) and nonfat milk in the target protein solution has been used to minimize the nonspecific interactions between peptides and target proteins. Moreover, an increase in ionic strength of an incubation solution by increasing the salt concentration also can reduce nonspecific interactions through the disturbance of electrostatic interactions between peptides and target proteins.
However, the inclusion of false-positive beads due to the interactions between labeled dyes on a protein and peptides is inevitable with screening by the fluorescence detection methods, including COPAS sorting. It is reported that the fluorescence patterns of sorted beads under a microscope appeared hollow for true positives and solid for false negatives. This was attributable to the difference in orientation between the peptide-protein interaction (true positive) and peptide-labeled dye (false negative) configuration. 23 However, the situation may be different according to the protein size and properties or the type of polymeric bead used for a peptide library. Moreover, the screening time was significantly increased by manual or automatic picking under a microscope.23,27 Therefore, we explored two strategies to minimize the inclusion of false-positive beads to make the COPAS sorting system more efficient and reliable.
First, sorting of possible false-positive beads from interactions between dyes on proteins and peptides on beads can be minimized by sorting fluorescent beads from an assay of a peptide library with Alexa Fluor 647–labeled protein above the maximum fluorescence level from an assay of the peptide library and Alexa Fluor 647–labeled lysine. Figure 5 shows one representative example. Second, all sorted objects in each well of a 96-well plate can be further analyzed to discard less meaningful beads based on the shape (broken beads and bubbles) or based on the profile of the uniformity of fluorescence along the length of a bead by Profiler software (Union Biometrica) before the next cleavage step. Additional actions before construction of a focused library can be taken to discard peptides with an excessive number (normally >2) of positively charged amino acids (R, K) and peptides without any motif decided by a homemade peptide sequence analysis program (PEPS) after a sequencing procedure.
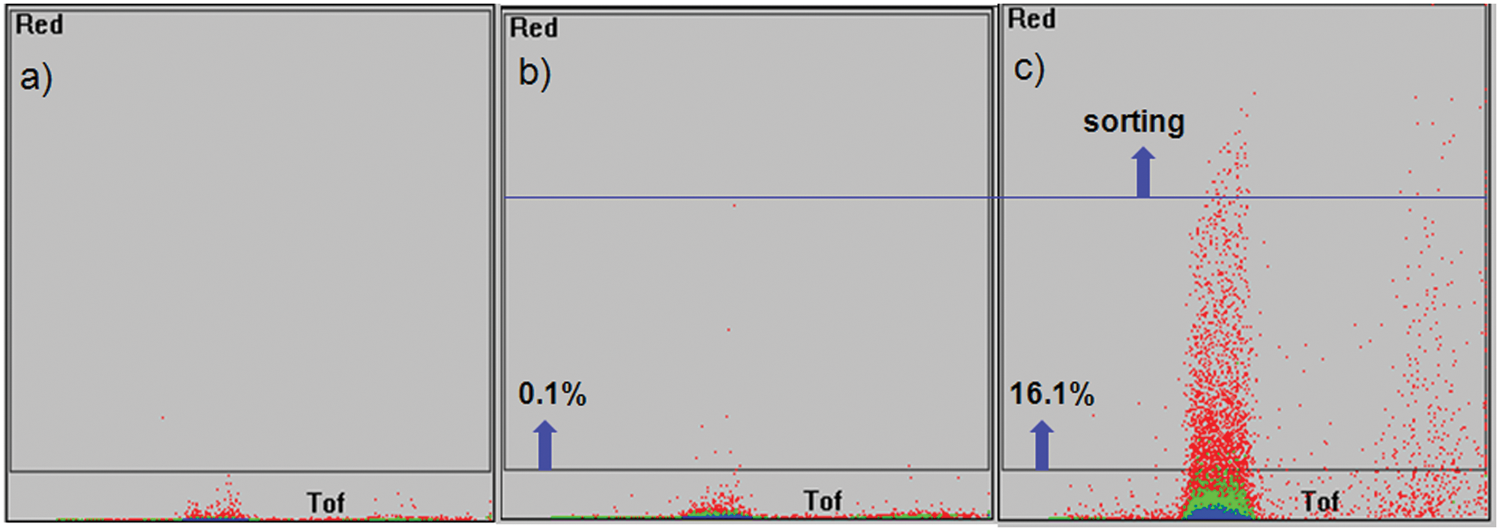
COPAS sorting images of (a) beads from a 5-mer peptide library, (b) beads from a 5-mer peptide library after incubation with Alexa Fluor 647–conjugated lysine (10 nM), and (c) beads from a 5-mer peptide library after incubation with a Alexa Fluor 647–labeled protein (10 nM) under the same sorting conditions.
The Profiler program (Union Biometrica) digitizes the object into a succession of peaks and valleys that directly traces the fluorescence intensity and displays subtle variations in fluorescence and extinction intensity (EXT, optical density) along the length of an object as it passes through the flow cell, including addressable sorted objects onto the 96-well plate. Using the visualized data by Profiler, bubbles, broken beads, and beads with an abnormally strong fluorescence profile (saturation of the detector) can be easily identified to exclude them for the following steps ( Fig. 6 ).
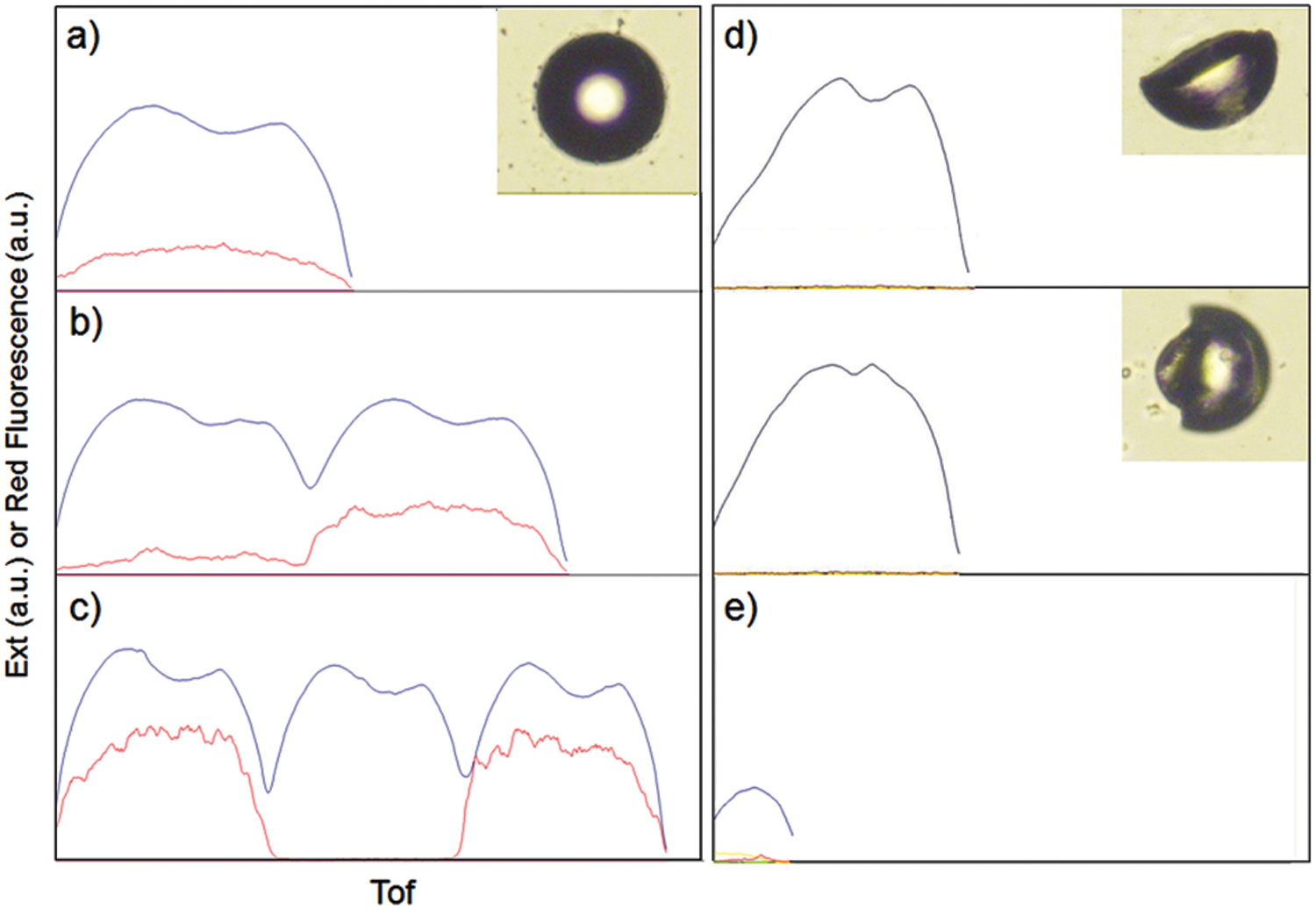
Profiler snapshots of (a) a singlet, (b) multiple beads (two beads), (c) multiple beads (more than two beads), (d) broken beads, and (e) a bubble in each well of a 96-well plate. The red line denotes the fluorescence intensity and the blue line denotes the extinction (EXT) along the length of an object.
Usually, broken TentaGel beads showed abnormally strong fluorescent intensity due to the exposure of the polystyrene core, resulting in a strong interaction with target proteins through hydrophobic interactions and the truncated EXT profile as well. Multiple beads in one well also could be identified by Profiler data ( Fig. 6b , c ). The identified multiple beads were transferred from the 96-well plates onto a glass slide. The slide was applied to a microarray scanner (GenePix; Molecular Devices, Sunnyvale, CA), and beads with strong fluorescence from the multiple beads were handpicked and placed individually back into the 96-well plates for the cleavage step.
PEPS generates several types of peptide sequence analysis results, such as (1) peptides with a particular matching pattern; (2) peptides with a defined pattern group, allowing for position shifting; (3) analysis of peptides via an alignment of a particular amino acid; and (4) histograms from the peptides. Figure 7 shows four output files of each analysis generated by PEPS obtained from one input containing one hundred 6-mer sequences. Based on the analysis, several approaches for the generation of focused libraries can be suggested. The generation of focused libraries by using peptides with motif sequences can lead to more efficient identification of peptide ligands with higher binding affinity and higher specificity. For example, a small-size focused library can be generated by using dominant amino acids at each position from peptides with a matching pattern, shown in Figure 7a .

Four types of peptide sequence analysis program (PEPS) output files resulting from one input file with 100 peptide sequences. (a) Sorting out peptides that contain a matching pattern (similar amino acids at three positions). The similar amino acids belong to the same group.* The exemplified output shows peptides with a hydrophobic amino acid in the fourth position, a hydrophobic amino acid in the third position, and a positively charged amino acid in the first position from the C-terminus. The matching pattern can be also extended to more than three positions. (b) Sorting out peptides that contain a defined pattern group, allowing for position shifting. (c) Output file by fishing out peptides that contain H. The sorted peptides are aligned with H located at position zero. Distribution of amino acids surrounding H from the aligned peptides that contain H is analyzed. (d) PEPS generates a table comprising the number of amino acid groups that occur depending on the position in the peptides (histogram). *The amino acids except G are bracketed into six groups (hydrophobic: A, V, L, I, F; positively charged: R, K, H; negatively charged: D, E; hydroxy: S, T; aromatic: Y, W, B; others: N, Q). B is used to designate phenylalanine for the aromatic group.
ultrafleXtreme MALDI-TOF/TOF for Automatic MS and MS/MS Acquisition and Semiautomatic Peptide Sequencing
For 5-mer or 6-mer peptides, de novo sequencing via MS/MS experiments is one of the best options to rapidly elucidate the peptide sequences. MS/MS-based sequencing offers great efficiency compared with other methods such as the conventional Edman degradation. In addition, modified amino acids are also allowed to be sequenced, which can be employed to give more structural diversity in the library. We have previously reported a novel and efficient high-throughput semiautomatic peptide sequencing algorithm used in combination with PEAKS software (Bioinformatics Solutions, Inc., Waterloo, Ontario, Canada). 28 Recently, MALDI-TOF/TOF instruments with much better sensitivity and resolution have been introduced. We observed significant improvement of signal to noise from Autoflex II to Ultraflex III ( Fig. 8 ). This improvement led to more accurate peptide sequencing through clearer identification of fragmented peaks. In addition, the FlexControl software supplied together with the MALDI-TOF/TOF allowed us to acquire MS and MS/MS data automatically. The obtained MS/MS data could be used for peptide sequencing after conversion to a data format compatible with PEAKS software.
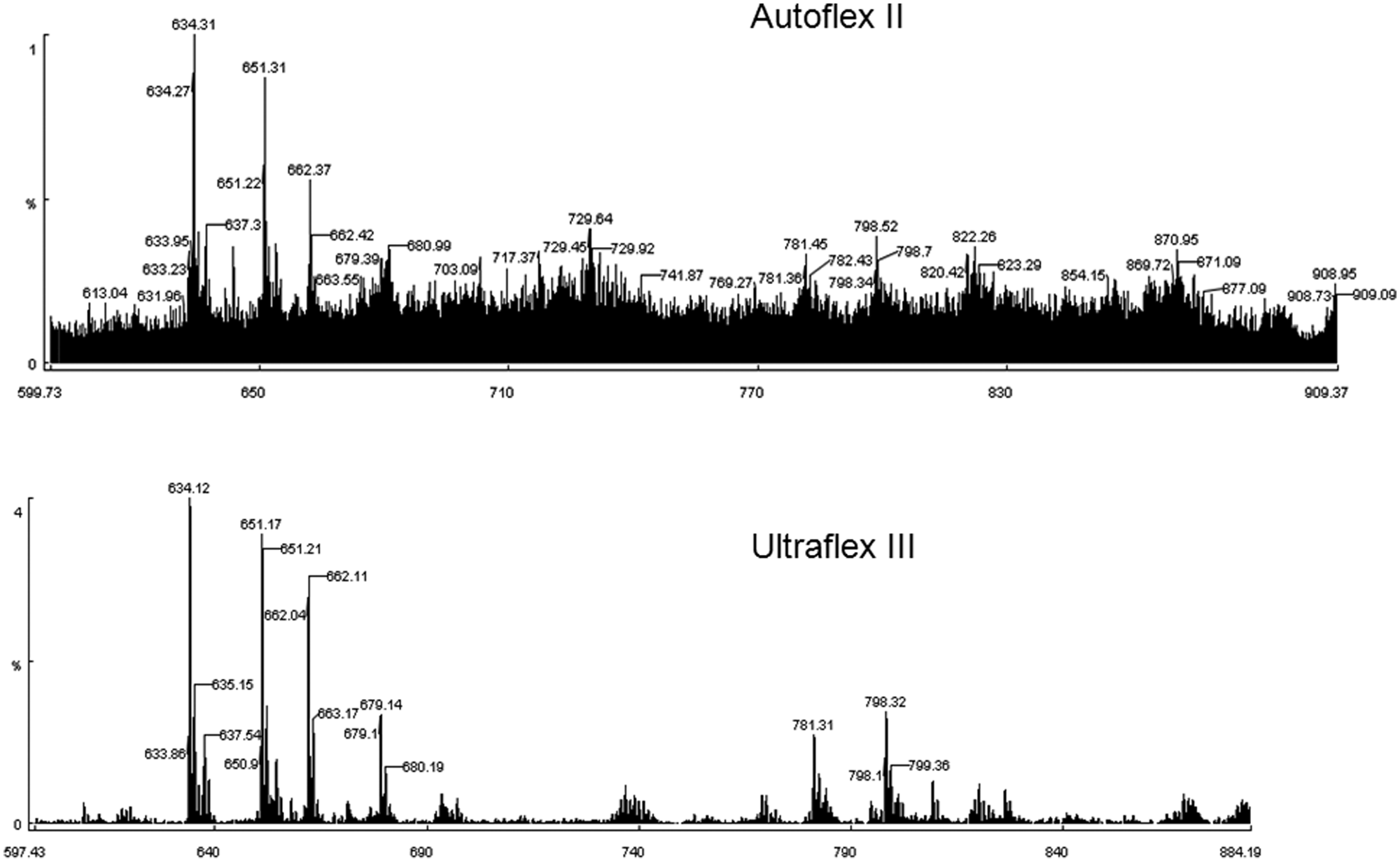
A comparison between Autoflex II and Ultraflex III. The tandem mass spectrometry (MS/MS) spectrum obtained by Ultraflex III gave a significantly higher signal-to-noise ratio from an identical peptide sample.
We have also developed a novel strategy to differentiate isobaric amino acids, such as I/L and K/Q, in our previous work. 28 With the use of the isobaric amino acid differentiation strategy coupled with PEAKS software, peptides are sequenced efficiently and precisely. Initially, a partial amount of G and A was added together with Q and I, respectively, at the coupling steps. However, the mass difference of 42 amu between I and A was not large enough to segregate the MS/MS peaks of fragmented peptides from the parent peptide due to the relatively strong intensity of the parent peaks. Therefore, G was selected to be paired with both Q and I for efficient encoding. The larger mass difference of 56 amu between I and G significantly helped in obtaining high-quality MS/MS spectra for facile and accurate sequencing ( Fig. 9 ).
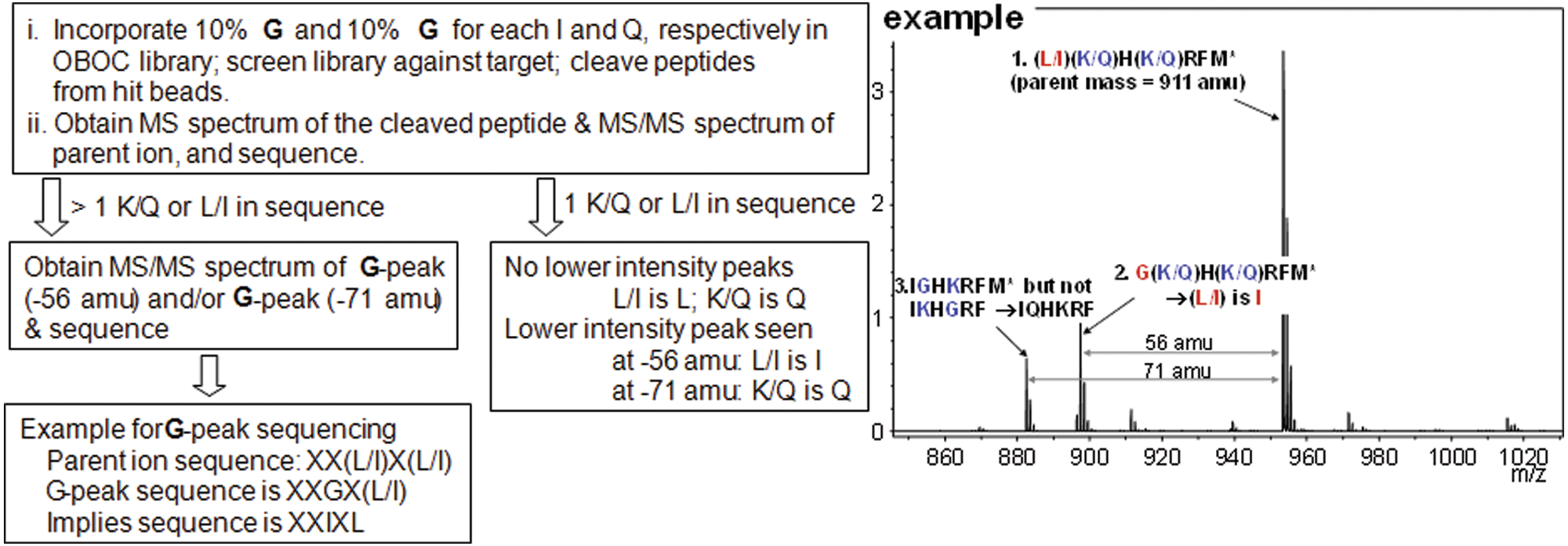
Procedure for differentiation of isobaric amino acids with one example. MS/MS, tandem mass spectrometry; OBOC, one-bead-one-compound.
The above automated or semiautomated steps were incorporated into the overall screening process, and all steps were smoothly linked to each other to make one-round screening done within 4 to 5 days ( Fig. 10 ).
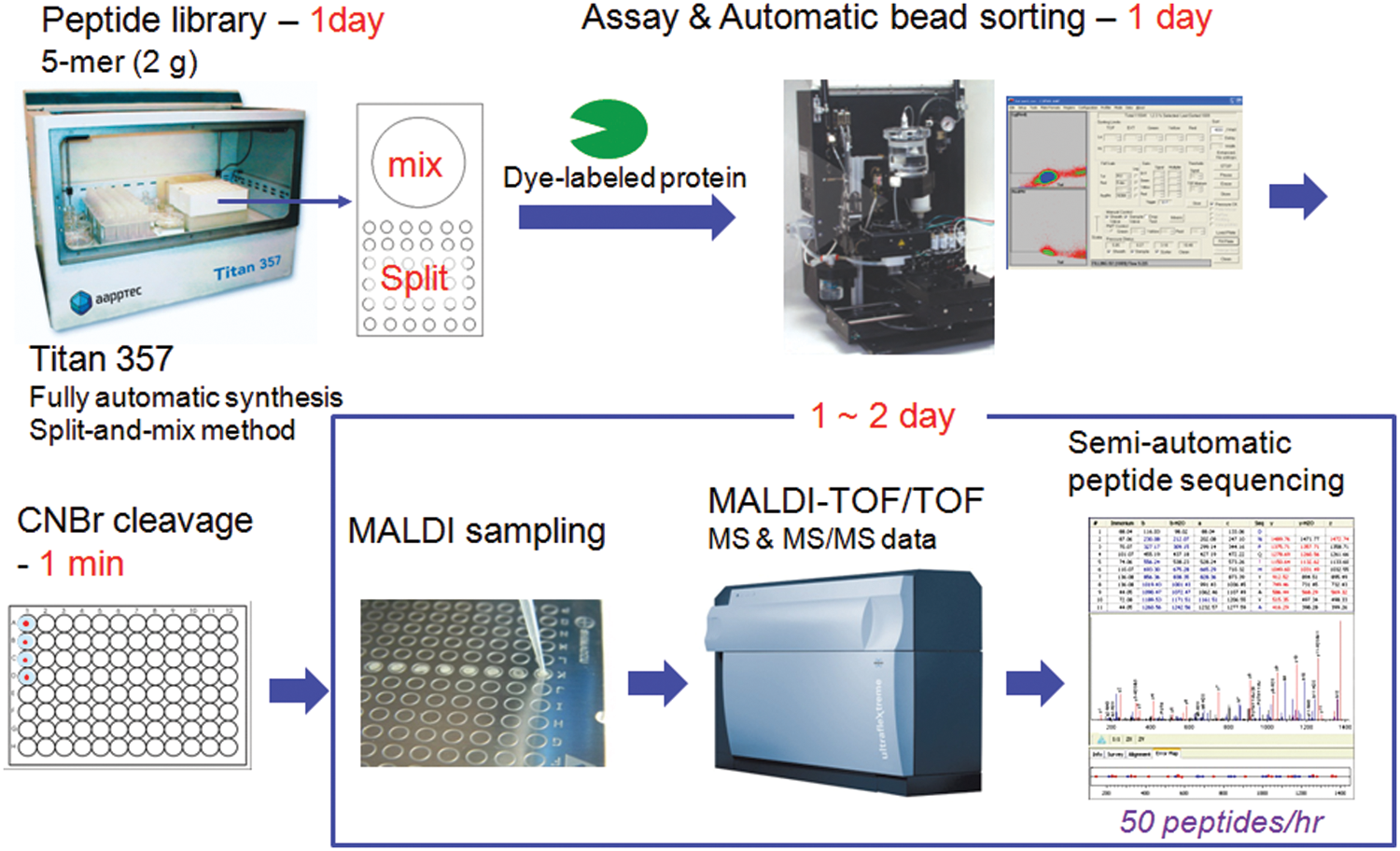
Overall screening process after automation and optimization. CNBr, cyanogen bromide; MS, mass spectrometry; MS/MS, tandem mass spectrometry; MALDI-TOF/TOF, matrix-assisted laser desorption ionization tandem time-of-flight.
Finding a 6-mer Peptide Ligand for Carcinoembryonic Antigen via the HTS Technology
To demonstrate how HTS works with a target protein, highly glycosylated carcinoembryonic antigen–cell adhesion molecule (CEACAM 5; Fitzgerald Industries International, Acton, MA) was screened with a comprehensive 6-mer peptide library that was constructed with Titan 357 and the isobaric differentiation strategy.
The CEACAM family contains at least 29 expressed proteins. Members of the CEACAM family belong to the immunoglobulin superfamily, with CEACAM 5 sharing several immunoglobulin domains. Namely, members of the CEACAM family contain 108 amino acids at the N-terminal homologous to the IgG variable domain, as well as up to six domains that are homologous to the IgG constant domain. The CEACAM family is expressed on the apical surface of epithelial cells and on squamous epithelial cells. Members of the CEACAM family exhibit a wide range of biological functions. Some common biological functions of the CEACAM family include environmental sensing via cell-cell adhesion, as well as the binding and uptake of infectious pathogens. CEACAM 1, for example, is also involved in the modulation of angiogenesis. Our primary target, CEACAM 5, plays a role in the promotion of metastasis and in “loss-of-contact” induced apoptosis. CEACAM 5 is a highly stable molecule, has almost negligible expression in healthy tissue, and has relatively high levels of expression in tumors of epithelial origin. This makes CEACAM 5 an excellent candidate as a clinical tumor marker. In addition to being used as a biomarker, carcinoembryonic antigen (CEA) levels are also being used to assess the effectiveness of treatment protocols and to determine the prognosis of colon cancer patients.29,30
A comprehensive 6-mer peptide library was generated by using D–amino acids and screened against Alexa Fluor 647–labeled CEACAM 5 to obtain 106 positive beads. A concentration of target protein was reduced to 10 nM to promote competition among the library beads. A solid laser (640 nm) with stronger power was used to sort positive beads instead of a diode laser used in our previous research.25,26 Most sequences of peptides obtained from the positive beads had a clear motif, such as wpxxxx, ypxxxx, or fpxxxx ( Table 1 ). A focused library was generated by using amino acids dominant at each position through analysis of positive peptides ( Fig. 11 ). Thirty-five positive peptides were identified from one-round stringent screening of the focused library under 5 nM of Alexa Fluor 647–labeled CEACAM 5 ( Table 2 ). Three peptide sequences appeared twice and were selected for validation by surface plasmon resonance (SPR). A 6-mer peptide ligand (wpykkw) among the three peptides showed the highest binding affinity by SPR experiments. It showed 8.2 µM of dissociation constant (Kd) calculated from steady-state affinity fitting of the SPR sensorgram using Biacore (GE Healthcare, Piscataway, NJ) T100 Evaluation software ( Fig. 12 ). The strong binding of wpykkw to CEACAM 5 was also confirmed by dot blot. In addition, wpykkw did not show any affinity for BSA used as a competing protein during assay and screening in dot blot.
One Hundred Six Positive Peptides from Screening of a Comprehensive 6-mer Library against CEACAM 5
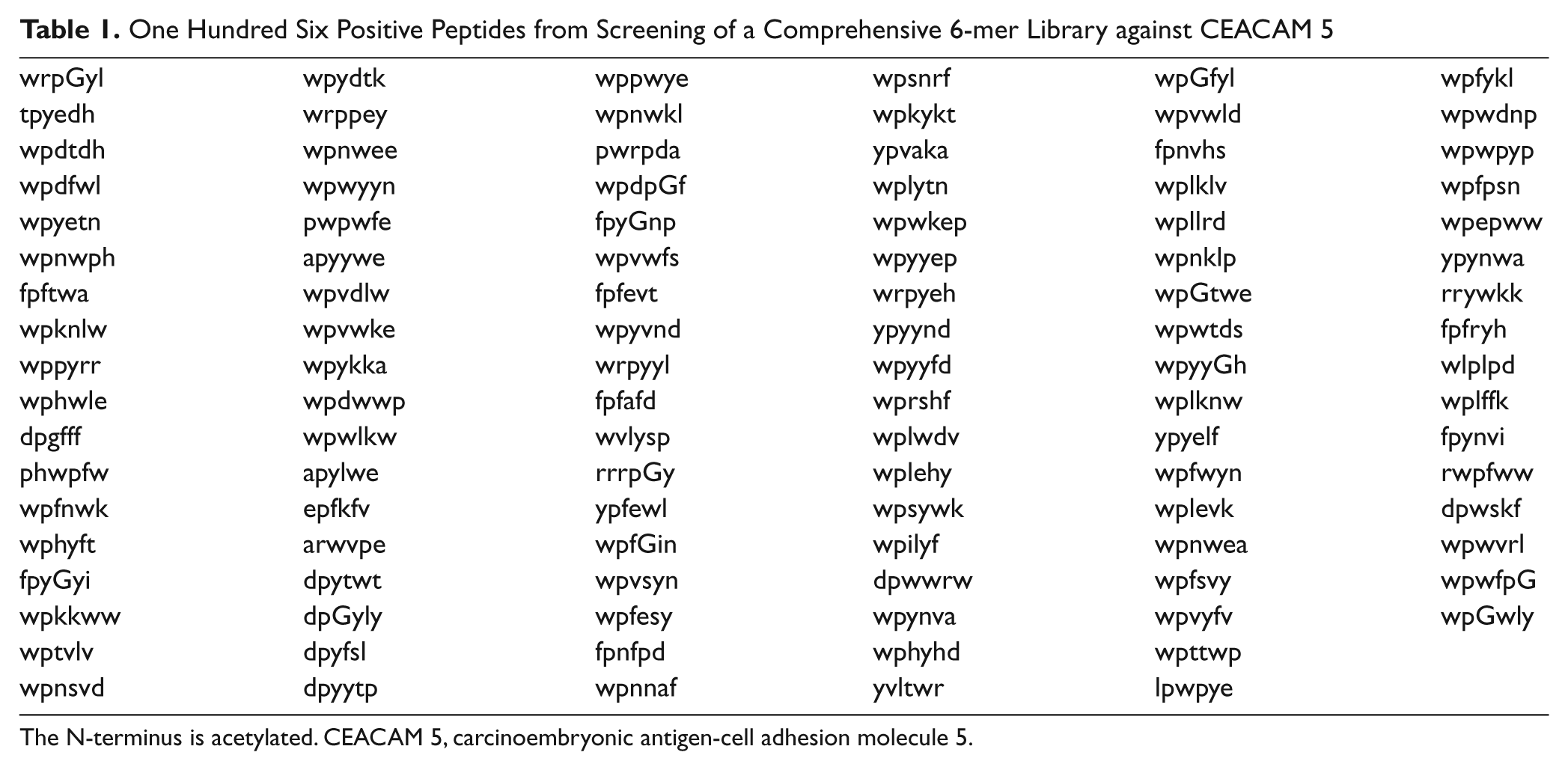
The N-terminus is acetylated. CEACAM 5, carcinoembryonic antigen-cell adhesion molecule 5.
Thirty-Five Positive Peptides from Screening of a Focused 6-mer Library against CEACAM 5
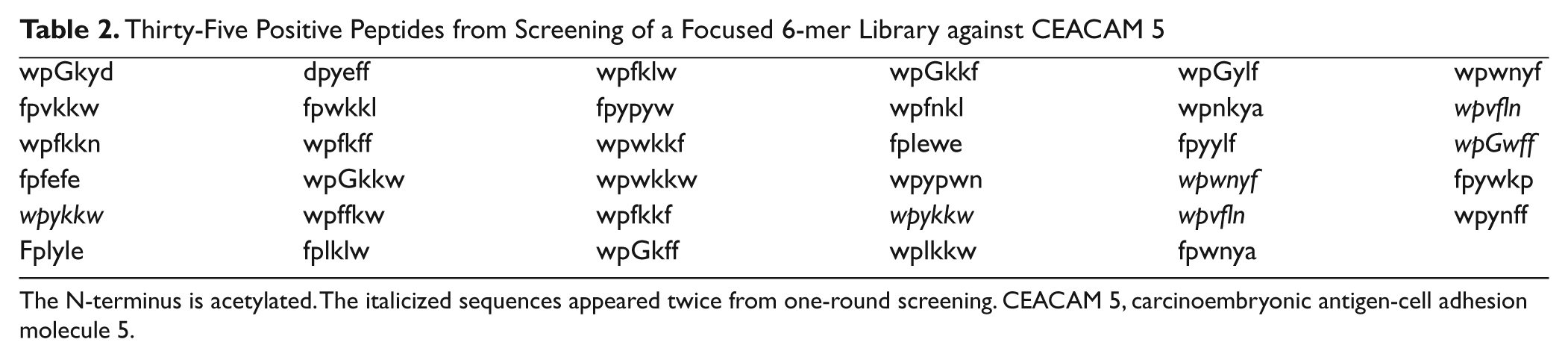
The N-terminus is acetylated. The italicized sequences appeared twice from one-round screening. CEACAM 5, carcinoembryonic antigen-cell adhesion molecule 5.
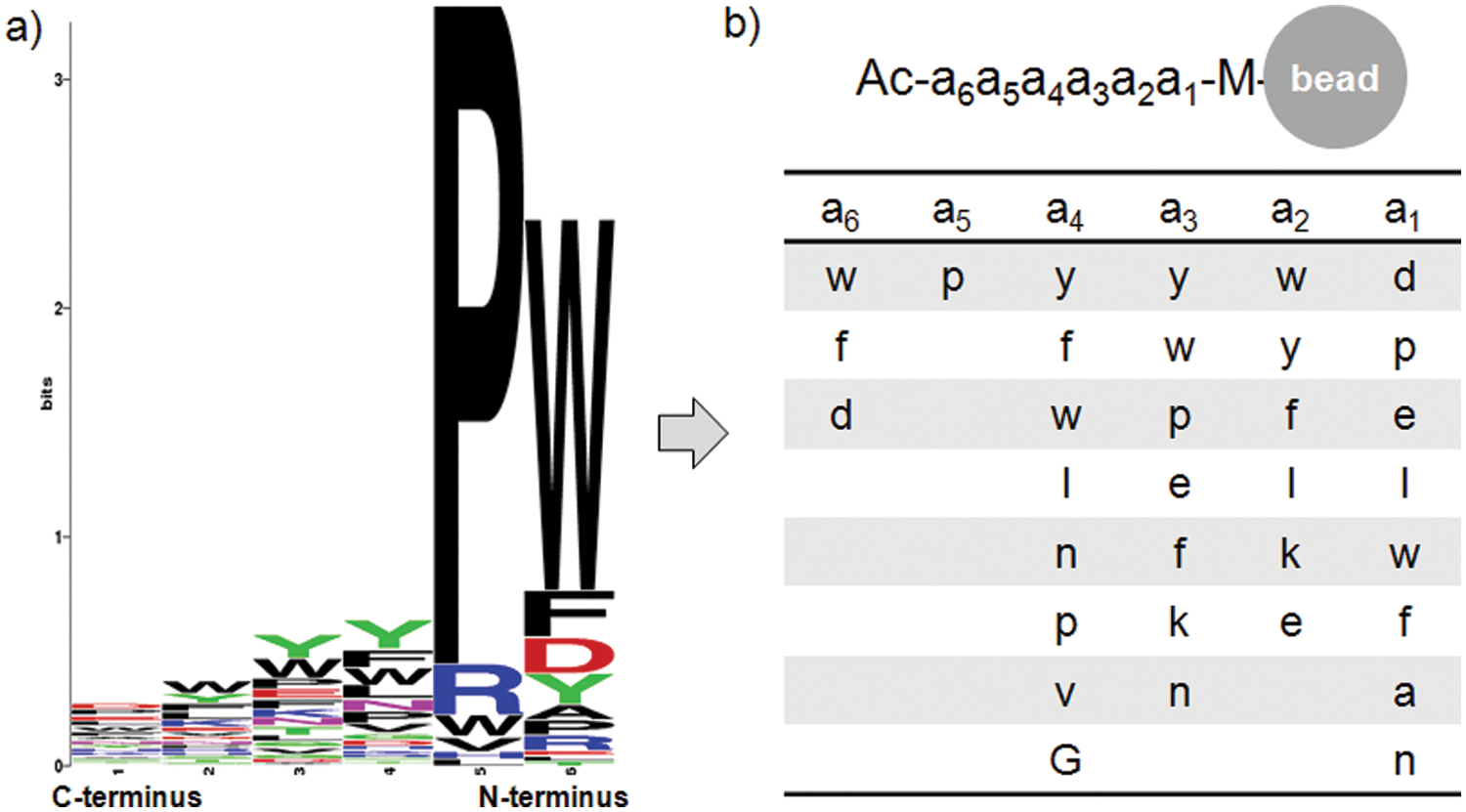
Generation of a focused library. (a) A font histogram (generated by a sequence logo generator [http://weblogo.berkeley.edu]) from the screening of a one-bead-one-compound (OBOC) peptide library against carcinoembryonic antigen–cell adhesion molecule 5 (CEACAM 5). (b) The structure of a focused library by using dominant amino acids at each position of the histogram.
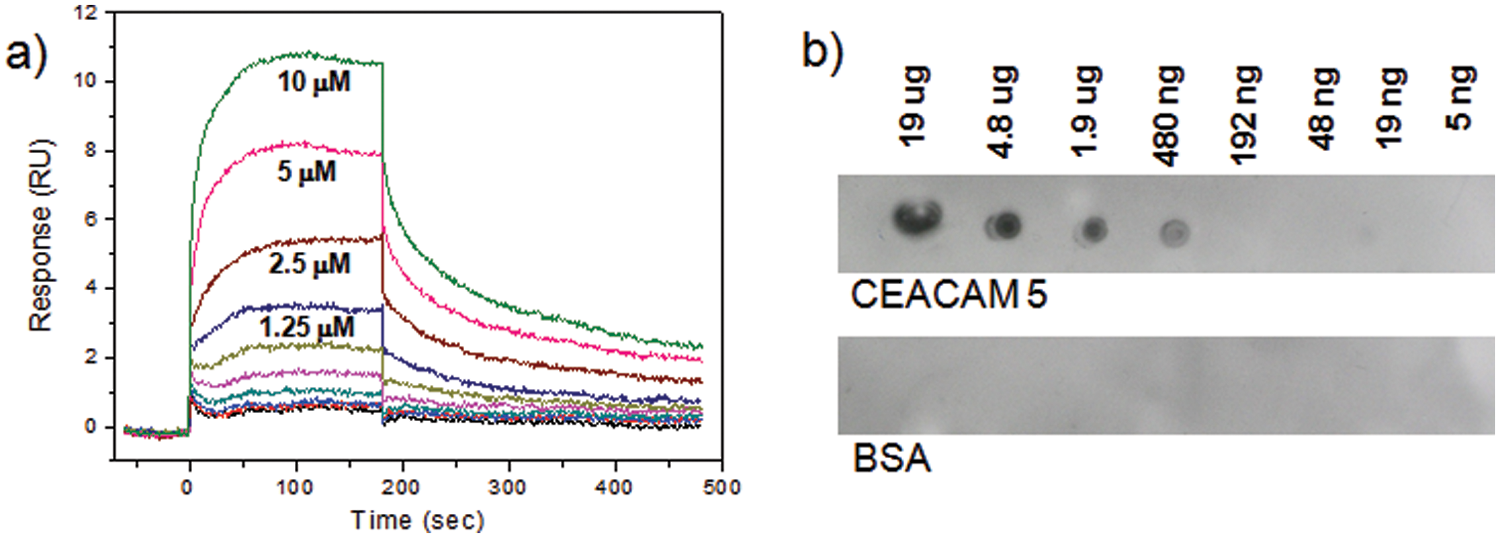
Validation of a peptide ligand (wpykkw) by (a) surface plasmon resonance (SPR; immobilization level = 650 response units [RU]) and (b) dot blot experiments ([peptide] = 100 nM). BSA, bovine serum albumin; CEACAM 5, carcinoembryonic antigen–cell adhesion molecule 5.
Conclusions
One round of screening of a 5- or 6-mer combinatorial peptide library could be completed within 4 to 5 days under a well-established, high-throughput platform. The ultra-HTS has been achieved by optimizing and automating time-consuming steps, which are the construction of a peptide library, the sorting of positive beads, and peptide sequencing via the acquisition of MS and MS/MS data. We have successfully demonstrated that three rounds of semiautomated screening resulted in the identification of a 6-mer peptide ligand for CEACAM 5 through the HTS process. Currently, we are exploring the full automation of the semiautomatic processes, including peptide library synthesis, de novo peptide sequencing, and MALDI-MS sampling to further facilitate the overall screening campaign.
Technical Description for Screening against CEACAM 5
General Procedures
NMP, diethylether, DCM, TFA, and Fmoc-NH-PEG2-CO2H (20 atoms) were purchased from Merck (Whitehouse Station, NJ). High-performance liquid chromatography (HPLC)–grade acetonitrile and water were obtained from J. T. Baker (Phillipsburg, NJ). Fmoc-protected D–amino acids and Rink amide AM resin (100–200 mesh, NH2 loading = 0.31 mmol/g) were obtained from GL Biochem (Shanghai, China). The pure peptides were obtained by purification using a preparative HPLC system (Gilson system) on a C18 reversed-phase preparative column (Kromasil from AkzoNobel [Amsterdam, the Netherlands], 5 µm, 250 × 30 mm). EZ-Link NHS-Biotin reagent was purchased from Thermo Scientific (Waltham, MA). Unless otherwise specified, chemicals were purchased from Sigma Aldrich (St. Louis, MO). MS and MS/MS data were obtained using a Bruker ultrafleXtreme MALDI-TOF/TOF. The PEAKS software was purchased from Bioinformatics Solutions, Inc. The microwave-assisted CNBr-mediated cleavage was performed using a household microwave oven (Model R-248J, 800 W, 2450 MHz) from Sharp, Inc. (Mahwah, NJ). CEACAM 5 was purchased from Fitzgerald Industries International.
Library Generation by an Automatic Peptide Synthesizer
Peptide libraries were synthesized by using an automatic synthesizer Titan 357 (AAPPTec). TentaGel S amino beads were swelled in NMP for 120 min in the collective vessel (CV). After draining the solvent, Fmoc-methionine (Fmoc-Met) was added to the CV, as well as TBTU and DIEA. The resulting suspension was vortexed for 30 min and washed repeatedly by NMP × 4, prior to the treatment of 20% piperidine in NMP for 5 min. The liquid was drained and a fresh aliquot of 20% piperidine in NMP was treated for 15 min. The resulting beads were thoroughly washed by NMP × 4 and DCM × 4, followed by distribution equally into 18 reaction vessels (RVs). Eighteen unnatural (i.e., D-) amino acids were employed as the diversity elements, excluding cysteine and methionine. Fmoc-isoleucine and Fmoc-glutamine contained 10 mol% of glycine each for discrimination of isobaric residues in the semiautomatic peptide sequencing. One of the amino acid diversity elements, TBTU and DIEA were added to each RV. After vortexing for 15 min, the coupling was repeated (double coupling). The resulting beads in each RV were washed by NMP × 4. Again, the Fmoc group was removed by a repeated treatment of 20% piperidine in NMP to each RV, as described above. Beads in each RV were thoroughly washed by NMP × 4 and DCM × 4 and then combined into CV. The overall split, coupling, deprotection, and mix processes were repeated until the beads appended a hexamer, including the initial methionine. The beads were transferred in a reactor equipped with a filter, and the protective groups in the residues were removed by treatment of TFA-water-TIS (94:3:3, v/v/v) for 2 h. The solvent was drained and the resulting beads were thoroughly washed by DCM × 3, methanol × 3, water × 3, methanol × 3, DCM × 3, and diethylether × 3, successively, and dried under reduced pressure.
Assay of Peptide Library against CEACAM 5
An Alexa Fluor 647 protein labeling kit (A20173; Invitrogen) was chosen as a source of dye for labeling CEACAM 5. Following the supplier’s protocol, 1 mg of CEACAM 5 labeled with a one-third amount of Alexa Fluor 647 from the kit was purified from the mixture solution by size exclusion purification resin included in the kit. The concentration of the purified dye-labeled CEACAM 5 was determined by UV-Vis spectroscopy (NanoDrop Technologies, Wilmington, DE). Then, 100 mg of library resin was transferred into an 8-mL Alltech vessel and preincubated in 2.5 mL of blocking buffer solution, 0.05% NaN3, 0.1% Tween 20, and 0.1% BSA in phosphate-buffered saline (PBS) buffer (pH 7.4), for 1 h on a 360° shaker at 25 °C. The buffer solution was drained, and then 2.5 mL of 10 nM dye-labeled CEACAM 5 in blocking buffer solution was added. The resulting mixture was incubated for 18 h at 25 °C. The liquid was drained and washed with blocking buffer × 3, 0.1% Tween 20 in PBS × 3, and PBS × 6.
Sorting by COPAS
All of the assayed library beads were transferred into a sample vessel of COPAS Plus (Union Biometrica) and diluted with 100 mL of 0.1% Tween 20 in PBS buffer. According to the two-stage sorting protocol described in the main text, positive beads were collected into one well (200 µL) of 96-well plates in an aliquot of 10 µL of water. Sorting parameters were optimized based on fluorescence signal of the bead, such as the gating region, defined as TOF versus red fluorescence of beads, and the sorting region selected positive beads based on TOF and uniformity red fluorescence (RedPH). The sorted beads were analyzed by the Profiler II software.
CNBr-Mediated Cleavage
Each bead immersed in 10 µL of water in each well of 96-well plates was thoroughly purged with nitrogen, before and after the addition of CNBr (10 µL, 0.50 M in 0.2 N HCl solution). The well plates were tightly sealed by a thin film on top and exposed under microwave conditions for 1 min. The resulting solution was concentrated under centrifugal vacuum for 30 min at 45 °C and then for 30 min at 60 °C.
Peptide Sequencing Based on MALDI-TOF/TOF Experiments
To each well of a 96-well plate, 10 µL of α-cyano-4-hydroxycinnamic acid (α-CHCA) solution prepared by dissolving 4 mg of CHCA in 1 mL of acetonitrile/water (70/30, v/v) was added, followed by the addition of 10 µL of acetonitrile/water (70/30, v/v) containing 0.1% trifluoroacetic acid. The well plate was centrifuged for a short time to locate the solution on the bottom of each well and sonicated for 5 min to completely dissolve the cleaved peptides. Then, the well plate was centrifuged for a short time to place the solution on the bottom of the well. The 2-µL aliquot of each peptide solution was taken up and spotted onto a 384-well MALDI sample plate. The resulting plate was stood to be air dried for 15 min. The MS spectrum of each peptide was acquired by an automatic acquisition method using the FlexControl software that operates Bruker’s ultrafleXtreme MALDI-TOF/TOF instrument. A parent mass peak from each MS spectrum was manually identified in the FlexAnalysis software and copied to a data sheet in FlexControl for automatic acquisition of the MS/MS spectrum. If there was a 56-amu smaller peak or a 71-amu smaller peak than the parent mass peak, its MS/MS spectrum was also obtained for isobaric differentiation. PEAKS software was used to elucidate the peptide sequences in a semiautomatic fashion with the acquired spectra.
Validation by SPR and Dot Blot
For the validation of positive peptides by dot blot, Biotin-NHS (Thermo Scientific) was used for bulk synthesis of C-terminal biotin-labeled ligands. Upon the completion of peptide construction as described in Figure 13 , the crude biotinylated ligand was precipitated with ether and then purified to give >98% in purity by C18 reversed-phase Prep-LC. Binding affinities were measured using the Biacore T100 system (GE Healthcare) equipped with research-grade CM5 sensor chips (GE Healthcare). The flow cell 1 (or 3) of the CM5 chip was used as a reference to subtract nonspecific binding, drift, and bulk refractive index, whereas the flow cell 2 (or 4) was immobilized with CEACAM 5. A response unit (RU) of 650 was reached for immobilization of CEACAM 5 onto the surface to the sensor chip by protocol of the thiol coupling kit. The binding of the peptide ligands to CEACAM 5 was also demonstrated through dot blot experiments in 0.5% nonfat dry milk in TBS-T (25 mM Tris, 150 mM NaCl, 2 mM KCl, 0.5% Tween-20 [pH 8.0]). A solution of CEACAM 5 was prepared as 2.4-mg/mL stocks in PBS buffer (pH 7.4) as per the protocol of the manufacturer. BSA solution was also prepared at the same concentration as CEACAM 5. A dilution series of CEACAM 5 and BSA solution was applied to a nitrocellulose membrane, typically ranging from 19 µg to 5 ng per spot. The 19-µg spot reached its concentration by adding five times of a 2-µL stock solution at the same spot after being completely dry. The membrane was treated in a blocking solution (5% nonfat milk/TBS-T) at room temperature for 2 h. The biotinylated peptide solutions were prepared at 100 nM concentration in 0.5% nonfat milk/TBS-T and incubated over the membrane for 2 h at ambient temperature. After washing with TBS-T for 10 min three times, 1:3000 Streptavidin-HRP (Abcam, Cambridge, MA) prepared in 0.5% milk/TBS-T was added to the membrane and incubated for another 2 h. After washing with TBS-T for 10 min three times, the membrane was treated to chemiluminescent reagents (Amersham ECL Plus Western Blotting Detection Reagents; GE Healthcare) and then immediately developed on film.
Synthesis of biotinylated peptides.
Footnotes
Acknowledgements
We thank Ms. Shi Yun Yeo for generous assistance and Prof. James R. Heath at Caltech for great advice.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).