
Abstract
A high-throughput label-free cell assay for modulating cell-cell communication is demonstrated with the Epic® system, a resonant waveguide grating sensor platform. Natural killer (NK) cells are known to be able to recognize abnormal cells (e.g., cancer cells and cells presenting intercellular adhesion molecule 1 [ICAM1] through cell surface receptors) and kill them. In this study, the effect of effecter cells NK92MI on two kinds of target cells, cervical cancer cells (HeLa) and Chinese hamster ovarian cells overexpressing ICAM1 (CHO-ICAM1), was examined. Living target cells’ response to NK92MI cells was monitored in real time and measured as wavelength shift in picometers. The authors showed that the detectability of target cell response is affected by multiple factors: the ratio of effecter cells to target cells (E/T), the interaction time of the two types of cells, and the target cell type. For example, with the effecter cells NK92MI and the same incubation time of 16 h, a minimal E/T ratio of 1 is required to detect HeLa cell response, whereas an E/T of 0.5 is sufficient to detect CHO-ICAM1 cell response. The authors confirmed that NK92MI cell–mediated target cell cytotoxicity results in negative optical signals and is associated with apoptosis mainly through caspase pathways. Distinct optical signals could be generated with the pretreatment of the target cells with various known pharmaceutical reagents, making the assay useful for discovering new chemicals that may affect cell-cell communications.
Cell-cell communication is essential throughout the life of an organism. In embryonic development, a cell’s migration to a specific location or differentiation into a particular cell type is largely instructed by neighboring cells. In an adult, the brain functions through interneuronal transmission, body movement depends on neuron-muscle interactions, the immune system works via lymphocytes’ ability to distinguish their own cells from foreign cells, and so on. Failure in normal cell-cell communication accounts for a variety of diseases (e.g., autoimmune disease and cancers).
The mechanism by which an effecter cell interacts with a target cell varies but can be classified into two large categories: one through a molecule released from an effecter cell (e.g., a neuron transmitter) and the other through a direct contact of two cells (e.g., the binding of a T cell to an antigen-presenting cell). Even though the knowledge about cell-cell communication is growing over the years, understanding its underlying mechanism in either normal or diseased conditions for various cell systems remains an important research subject.
Cell-mediated cytotoxicity is one of the systems most extensively studied, as it plays an important role in the immune defense system and presents a promising means for cancer therapy. It often requires direct cell-cell contact, which is mediated through the recognition of receptors and ligands present on the surfaces of effecter and target cells.1–4 Many technologies have been developed to measure target cell death triggered by an effecter cell. Biochemical assays typically involve protein extraction and substrate labeling. Most cell-based assays require either radio or fluorescent labels. The release of a labeled compound preloaded inside of a target cell, the integrity of labeled DNA retained in a target cell, or the detection of caspases of a labeled target cell is measured and quantified.1,2,5–8 Recently, deploying electrical impedance measurements for assessing cell-mediated cytotoxicity has been demonstrated but in a medium-throughput format (96-well plate) and a different label-free technology.9,10
A resonant waveguide grating (RWG) sensor is able to measure reflective index change as a result of mass change near the sensing surface, usually ~120 to 200 nm in depth. Even though a mammalian cell typically has a height of >500 nm, its dynamic mass redistribution (DMR) in response to a stimulus can be detected and quantified with an RWG sensor if the DMR involves mass increase or decrease at the bottom portion of the cell perpendicular to the sensing surface. 11 A unique optical signature could be generated by monitoring DMR for a given cell type stimulated with a particular stimulus in a defined environment. This optical signature is robust and reproducible, and its usage as a novel measure for probing various cellular events has been intensively explored.11–13
In this study, we explored the use of an RWG optical sensor, Corning Epic System, for monitoring cell-cell communications in a high-throughput fashion. Among the many experimental model systems, we chose a cell-mediated cytotoxicity model system consisting of natural killer (NK) cells as effecter cells and two kinds of target cells: (1) Chinese hamster ovarian (CHO) cells overexpressing intercellular adhesion molecule 1 (CHO-ICAM1) and (2) cervical cancer HeLa cells because of the extensive knowledge available from the literature. NK92 is a highly cytotoxic NK cell line that is commonly used as a model system for evaluation of NK cell function. NK-92MI is an interleukin-2 independent NK cell line derived from the NK92 and is cytotoxic to a wide range of malignant cells. Both NK-92 and its derivative cell line NK-92MI have been intensively characterized and have the positive surface marker for CD2, CD7, CD11a, CD28, CD45, CD54 (ICAM-1), and CD56 but negative surface marker for CD1, CD3, CD4, CD5, CD8, CD10, CD14, CD16, CD19, CD20, CD23, CD34, and HLA-DR. Similarly, CHO cells are frequently used in studies of genetics, toxicity screening, and gene expression. CHO-ICAM1 cell line is an engineered CHO cell line that stably expresses a high level of human ICAM1, which is a surface marker recognized by NK92MI cells. Therefore, the CHO-ICAM1 cell line provides a good model for the study of the interaction between NK cells and ICAM1-related cells. Of many cancer cell models, the HeLa cell line is one of the oldest and most commonly used human cell lines for research related to cancer, AIDS, effects of radiation and toxic substances, gene mapping, and many other scientific pursuits. Hence, the HeLa cell line is a good model for addressing the interaction of NK cells and human carcinoma cells.
Using the two model systems, we demonstrate that the Epic system can robustly detect DMR changes of a target cell triggered by an effecter cell. With further studies using modulators involved in various cellular functions, we are able to correlate negative DMR signals with the cytotoxicity of the effecter cells, suggesting that DMR signals may be used as a quantitative measure for interrogating the mechanism of cell-mediated cytotoxicity. With its ability to monitor target cell DMR response to an effecter cell in real time and without the need to incorporate labels before or after the assay, the Epic system represents a powerful new tool for study of cell-cell communications and for discovery of drugs that interfere with the communication. To our knowledge, this is the first demonstration of measuring cell-cell interactions in a high-throughput label-free setup.
Materials and Methods
Materials
NK cells NK92MI, HeLa cells, parental CHO cells CHO-K1, engineered CHO-K1 overexpressing ICAM1 receptor (CHO-ICAM1), horse serum, fetal bovine serum (FBS), EMEM, and RPMI-1640 (RPMI) were from American Type Culture Collection (ATCC, Manassas, VA). Dulbecco’s phosphate-buffered saline (PBS) was from Invitrogen (Carlsbad, CA).
Chemical brefeldin A (BFA) was from Tocris (St. Louis, MO); camptothecin and vinblastine were from Sigma Chemical Co. (St. Louis, MO). BFA is a fungal metabolite and primarily used as a biological research tool for studying protein transport. Treatment with BFA leads to a rapid accumulation of proteins within the endoplasmic reticulum (ER) and collapse of the Golgi stacks, and prolonged exposure can induce apoptosis. Camptothecin is a cytotoxic quinoline alkaloid that inhibits the DNA enzyme topoisomerase I and prevents DNA relegation, therefore causing DNA damage that in turn results in apoptosis. Vinblastine is an antimicrotubule drug used to treat certain cancers, including Hodgkin’s lymphoma, non–small-cell lung cancer, breast cancer, head and neck cancer, and testicular cancer. It interrupts microtubules and can induce apoptosis in cultured hepatocytes and human lymphoma cells. AFC fluorescently labeled peptides and their corresponding fluoromethyl keton (FMK) derived peptides for caspases 2, 3, 6, 8, and 9 were purchased from three vendors. Ac-DEVD-AFC/-FMK for caspase 3, Ac-VEID-AFC/-FMK for caspase 6, and Ac-IETD-AFC/-FMK for caspase 8, and Z-LEHD-FMK were from BD Biosciences (San Jose, CA); Z-VDVAD-AFC for caspase 2 and Ac-LEHD-AFC for caspase 9 were from EMD Biosciences (San Diego, CA). Z-VDVAD-FMK for caspase 2 was from R& D Systems (Minneapolis, MN).
Epic 384-well microplates have a sensor (waveguide grating) at the bottom of each well and were purchased from Corning Inc. (Corning, NY). Each plate was cleaned by exposure to high-intensity ultraviolet light, UVO cleaner made by Jelight Company Inc. (Laguna Hills, CA), for 6 min prior to use.
Cell Culture
NK-92MI cells were grown in RPMI medium supplemented with 12.5% horse serum (v/v), 12.5% fetal bovine serum (FBS) (v/v), and 1% antibiotics. CHO-K1 cells and CHO-ICAM1 cells were grown in RPMI medium supplemented with 10% FBS and 1% antibiotics. HeLa cells were grown in EMEM medium supplemented with 10% FBS (v/v) and 1% antibiotics. Cell number was counted with Beckman Coulter Particle Count (Beckman Coulter, Fullerton, CA).
Corning Epic System
The Corning Epic System, a wavelength interrogation system with transverse magnetic or p-polarized TM0 mode, was used in this study. The local reflective index change is measured as wavelength shift, typically in picometers (pm), which is directly proportional to the mass change within the sensing area, ~150 to 200 nm perpendicular to the surface. Baseline was recorded for approximately10 min when target cells were treated with an assay solution. The cell response signal was recorded immediately after a compound or an effecter cell was added to the monolayer target cells on the sensor. Relative to the baseline, an increase or decrease in mass in response to an analyte (a compound or an effecter cell) is measured as positive (+) pm or negative (–) pm.
Epic Cell-Cell Communication Assays
The cell-cell communication assay involves two types of cells: target cells and effecter cells, which were prepared separately. Approximately 1 to 2 × 104 target cells of CHO-K1, CHO-ICAM1, or HeLa in 50 µL medium containing 10% FBS were dispensed into each well of an Epic 384-well microplate and cultured at 37 °C in a humidified 5% CO2 incubator until ~90% of confluence (2–4 × 104 cells/well) was reached (~24 h). After removal of the growth medium for the target cells, 40 µL of RPMI medium containing 2% FBS and 1% antibiotics was added to each well. In parallel, effecter cells NK92MI were collected by spinning at 800 g for 10 min, washed with PBS, and resuspended in an appropriate volume of RPMI medium containing 2% FBS based on cell count to allow for the interaction of effecter cells and target cells at the ratio of 0.5 (E/T) unless stated otherwise. Twenty microliters of the effecter cell suspension was aliquoted into each well of a regular 384-well microplate (Corning Inc.).
The plates of both the target cells and the effecter cells were incubated in the Epic system for at least 30 min until the temperature reached 28 °C. Baseline signal of the target cells was recorded for 10 min. Quickly and carefully, effecter cells were transferred to the Epic 384-well microplate with the target cells already grown. Target cell response measured as a wavelength picometer (pm) shift was monitored over night.
To study the effect of pharmacological agents, target cells CHO-ICAM1 had been pretreated with an agent for 1 h unless stated otherwise before effecter cells NK92MI were introduced. To minimize the effect of temperature fluctuation and evaporative cooling, all studies were carried out at 28 °C with a lid on the microplate except for the time (~seconds) when a solution was introduced.
Conventional Caspase Enzymatic Activity Assay
Caspase assay for the enzymatic activity of caspase 2, 3, 6, 8, and 9 was performed using cell lysate following the manufacturer’s instruction. Briefly, a target cell lysate from the cells treated with a compound followed by NK cells was incubated with an AFC-labeled substrate specific to a caspase. The enzymatic activity of the caspase was quantified by the level of free AFC cleaved from the labeled substrate, which emits a yellowish green fluorescence and could be detected with a plate reader at ~505 nm. AFC release was monitored in a PerkinElmer Victor X4 Multilabel Reader at an excitation wavelength of 400 nm and an emission wavelength ranging from 480 nm to 520 nm. The specificity of the enzymatic reaction was determined by addition of excess corresponding FMK-derived substrates, which prevent the cleavage of the labeled substrates.
Results and Discussion
Effect of NK92MI Cells on HeLa Cells
To the monolayer target HeLa cells, effecter NK92MI cells were introduced at E/T ratios of 0 (medium only), 0.5, 1, 2, 4, and 8. The HeLa cell response was continuously monitored over a 20-h period. The observed picometer shifts (pm) at a given E/T ratio were captured at 1-h intervals and plotted as shown in Figure 1 . Clearly, a continuous decrease of pm (negative DMR signals) over the time was observed when the E/T ratio was 1 or higher ( Fig. 1 ), suggesting that there was loss of mass within the sensing region. The extent of mass loss was affected by not only the E/T ratio but also the incubation time for the two types of cells to interact.
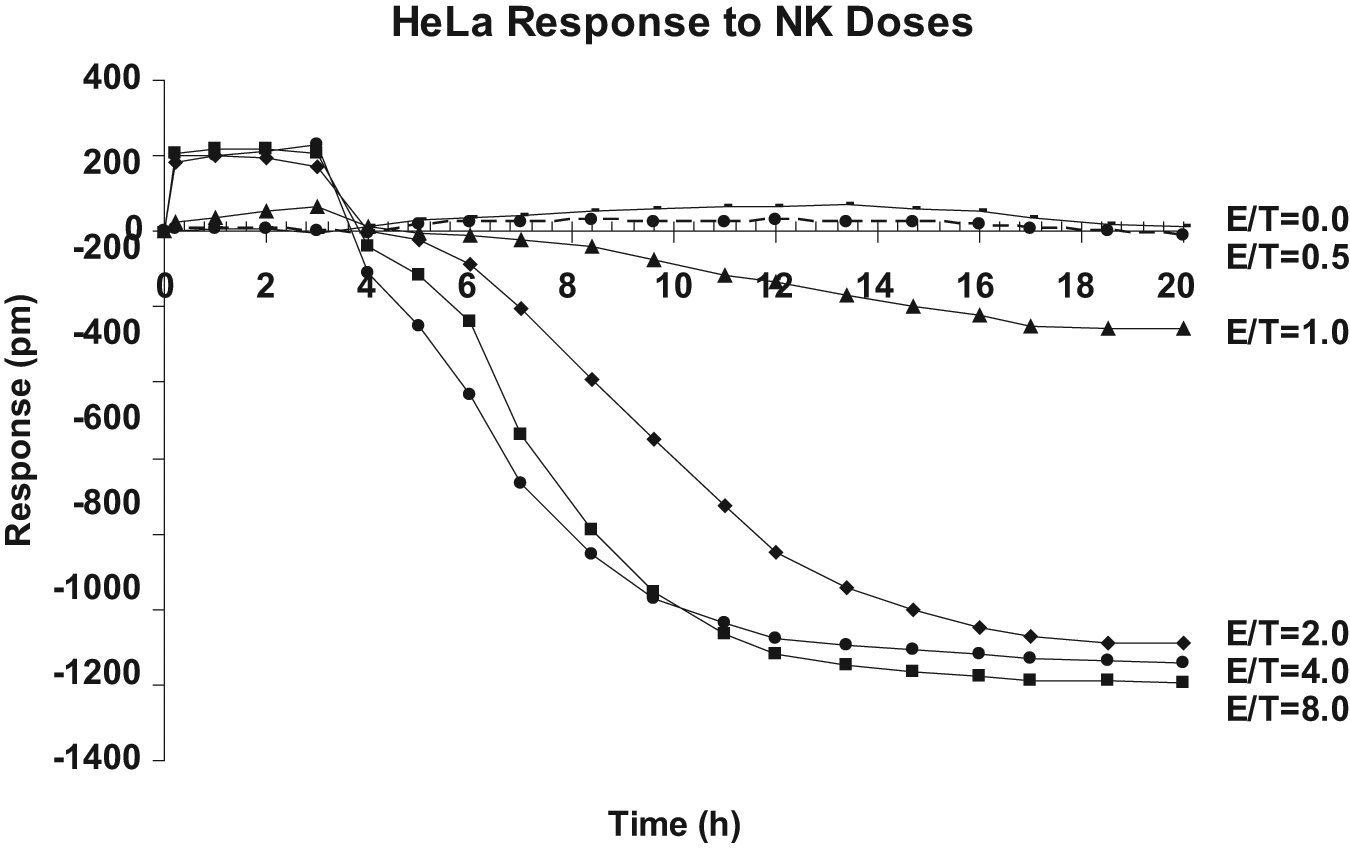
Time course of NK92MI cell-mediated cytotoxicity of HeLa cells at various effecter cells to target cells (E/T) ratios. In each well of a 384-well Epic microplate, 15 × 103 HeLa cells were seeded for ~24 h until reaching >90% confluence (with ~30 × 103 cells). In a regular 384-well microplate, 20 µL of medium containing various amounts of effecter NK92MI cells was prepared, each in four replicates, and carefully added to individual wells with confluent HeLa cells to achieve an E/T ratio of 0 (medium alone), 0.5, 1, 2, 4, and 8, respectively. The Epic microplate containing mixed cells was sent into the Epic system and continuously monitored for picometer (pm) shift at 1-h intervals for 20 h. HeLa cell response in pm at a given E/T ratio was averaged and plotted. An E/T ratio of 0 (target cells alone) served as the reference point. Baseline reading was recorded for the first 10 min of the assay, just before the NK92MI cell addition.
As expected, there was a reverse correlation between the E/T ratio and the time required to detect the target cell response. With increased E/T ratios from 1 to 2, 4 and 8, the time needed to show greater than −200 pm was significantly reduced from 17 h to 7, 6, and <5 h, respectively. In this system, a minimal E/T ratio of 1 appeared to be required for detecting any changes triggered by NK92MI cells. Moreover, at this E/T ratio, the maximum shift was reached after 17 h and remained small, approximately −200 pm. However, at an E/T ratio of 2 or higher, the HeLa cell response was not only faster but also bigger, reaching a maximum of approximately −1200 pm regardless of the E/T ratio. In other words, at the E/T ratio of 2 or higher, more effecter cells appeared to affect only the time required to reach maximum mass loss.
The maximum mass loss at the E/T ratios of 2, 4, or 8 reached plateau after 16 h of incubation. Taking the endpoint measurement after 16 h of incubation, a clear dose-dependent response of HeLa cells to NK92MI cells was observed ( Fig. 2 ). With an increasing ratio of NK92MI to HeLa, the response of HeLa was increased whereas the control CHO-K1 remained unchanged ( Fig. 2 ).
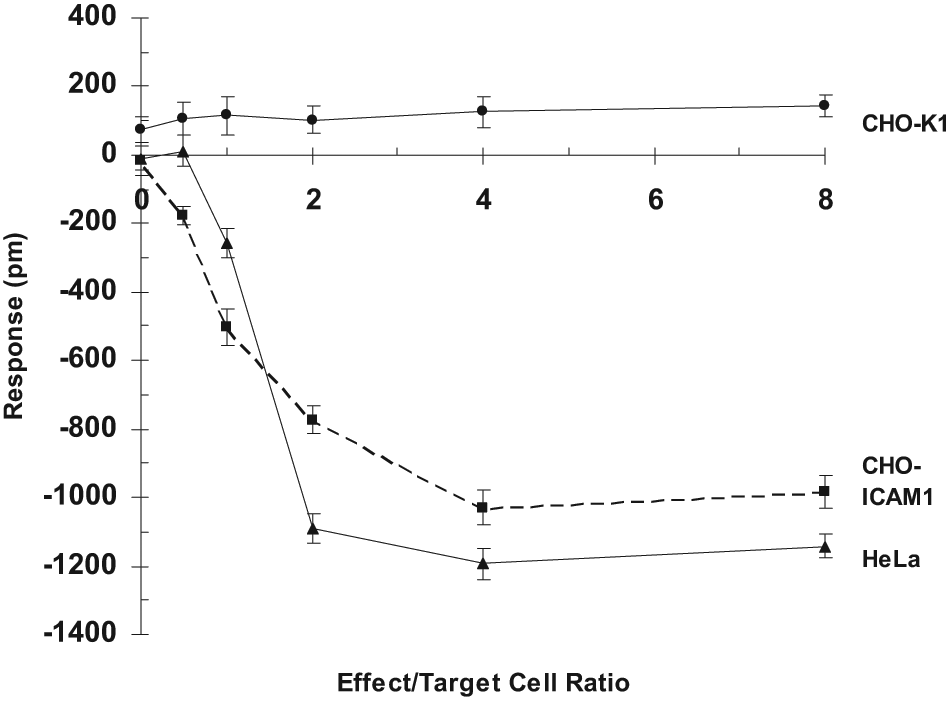
Dose-dependent effect of NK92MI cells. In each well of a 384-well Epic microplate, 15 × 103 of target CHO-K1, CHO-ICAM1, or HeLa cells were seeded for ~24 h until reaching >90% confluence (with ~30 × 103 cells). In a regular 384-well microplate, NK92MI cells were serially diluted threefold. Individual dilutions, each in four replicates, were added to the Epic microplate carrying confluent CHO-ICAM1 cells, HeLa, or CHO-K1 cells (negative control) to achieve effecter cells to target cells (E/T) ratios from 0 to 8. A single endpoint picometer (pm) shift was measured after 16 h incubation. The target cell response in pm was plotted.
Our results are in a good agreement with the literature. Researchers have found that NK cells are a type of lymphocyte and are already specialized to kill certain types of target cells via apoptosis. Morphologically, apoptosis is first characterized by a change in the refractive index of the cell, followed by cytoplasmic shrinkage and nuclear condensation and later the appearance of blebs and spikes of the cell membrane. 14 All these changes may translate to a decrease in the target cell mass within the sensing region and can be readily detected by the Epic system as a negative signal.
NK cells are known to kill cancer cells and target cells infected with virus.15,16 HeLa cells are cancer cells and have been reported to contain human papilloma virus 18; therefore, they could be killed by NK cells. Our observation of the correlation between negative DMR signals and an increasing amount of effecter cells suggested that NK92MI cells could induce negative DMR responses of HeLa cells, which is indicative of NK92MI cell-mediated cell death. Viral infection in mammalian cells is common in diseases such as AIDS, influenza, and hepatitis. Early detection and/or elimination of infected cells are critical for the control of these diseases. This study suggested that it is feasible to use the Epic system for screening chemicals that may affect NK cell–mediated cytotoxicity of cancer cells.
Of note is that at the first 3 h and at an E/T ratio higher than 1, there were small but clearly noticeable positive DMR signals. It may be caused by the initial mass increase contributed by the effecter cells before their ability to trigger target cell death took an effect.
Effect of NK92MI Cells on CHO-ICAM1 Cells
To see whether the optical signal of NK cell–mediated cytotoxicity is general for different types of target cells, a new target cell type, CHO-ICAM1 cells, was studied. A series of 10-fold dilution of NK92MI cells were first prepared, and individual dilutions were then added to a fixed amount of CHO-ICAM1 cells grown in a confluent monolayer, achieving various E/T ratios in the range of 0 to 8. The time course at the various E/T ratios was then plotted. Unlike HeLa cells, the maximum response of CHO-ICAM1 cells remained distinct at a given E/T ratio even after 16 h of incubation ( Fig. 3 ). Taking the endpoint measurement after 16 h of incubation, a clear dose-dependent response of CHO-ICAM1 cells to NK92MI cells was observed ( Fig. 2 ). With increasing ratio of NK92MI to CHO-ICAM1, the response of CHO-ICAM1 was increased whereas parental CHO-K1 remained unchanged ( Fig. 2 ). As expected, NK92MI cells triggered a negative signal indicative of the decrease in cell mass near the sensing region as a result of apoptosis of the CHO-ICAM1 cells. The initial response of approximately −200 pm was observed when the E/T ratio was 0.5. As the ratio increased, the negative response of CHO-ICAM1 was further increased until hitting the plateau of approximately −1000 at the E/T ratio of 8. The fact that control CHO-K1 cells, which lack the ICAM1 receptor on the cellular surface, were not responsive to an increasing amount of NK92MI cells suggested that the Epic cell response was specific and was mediated through the interaction of NK92MI to the ICAM1 receptor presented on CHO cells.
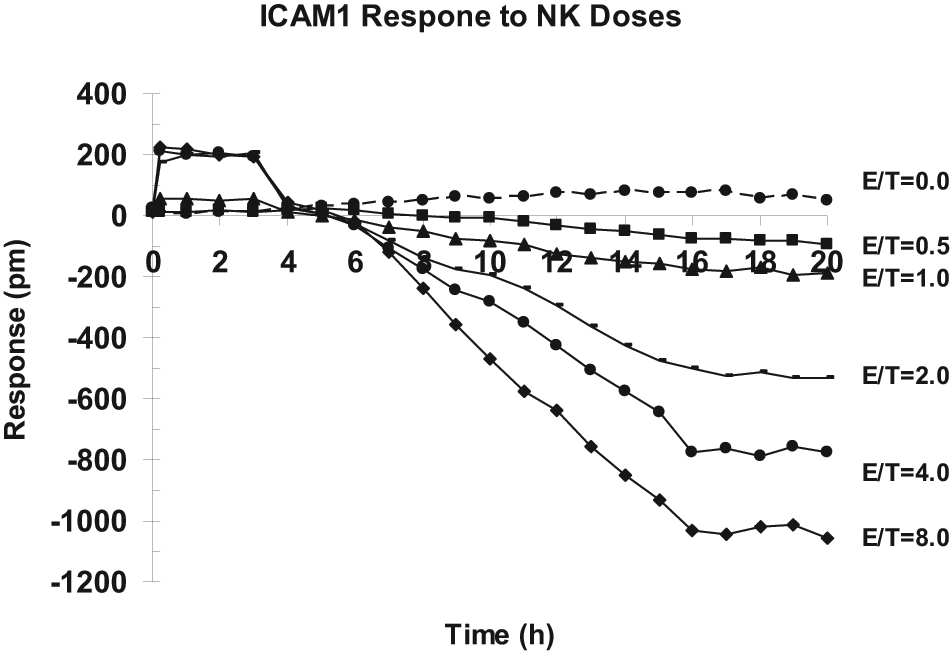
Time course of NK92MI cell–mediated cytotoxicity of CHO-ICAM1 cells at various effecter cells to target cells (E/T) ratios. In each well of a 384-well Epic microplate, 15 × 103 CHO-ICAM1 cells were seeded for ~24 h until reaching >90% confluence (with ~30 × 103 cells). In a regular 384-well microplate, 20 µL of medium containing various amounts of effecter NK92MI cells was prepared, each in four replicates, and carefully added to individual wells with confluent CHO-ICAM1 cells to achieve an E/T ratio of 0 (medium alone), 0.5, 1, 2, 4, and 8, respectively. The Epic microplate containing mixed cells was sent into the Epic system and continuously monitored for picometer (pm) shift at 1-h intervals for 20 h. CHO-ICAM1 cell response in pm at a given E/T ratio was averaged and plotted. An E/T ratio of 0 (target cells alone) served as the reference point. Baseline reading was recorded for the first 10 min of the assay, just before the NK92MI cell addition.
The observation is consistent with the literature showing that NK92 can recognize and subsequently cause the death of ICAM1-presenting cells.17,18 It was found that increased expression of ICAM1 accounted for the increased sensitivity of neuroblastoma to lysis by NK cells. 19 The NK92MI and CHO-ICAM1 system seemed to be more sensitive as it requires an E/T ratio of only 0.5 to see a clearly noticeable effect, approximately −200 pm; therefore, it was chosen for further studies.
Effect of Modulators on Effecter Cell–Induced Apoptosis of Target Cells
To comprehend the mechanism leading to the negative DMR signal observed in the aforementioned studies, the impact of a number of modulators on NK cell–mediated apoptosis was examined. The target CHO-ICAM1 cells were pretreated with caspase 3 inhibitor Z-DEVD-FMK (Cas3In), camptothecin, vinblastine, and BFA individually for 1 h and then challenged with NK92MI cells at the E/T ratio of 0.5 for 16 h. We choose the E/T ratio of 0.5 to see whether a compound has a significant effect on the target cells because at high E/T ratios, the large impact of effecter cells on the target cells would mask the effect of a compound. The results of CHO-ICAM1 cell response are shown in Figure 4 . The pretreatment of the caspase 3 inhibitor, even at its lowest concentration of 0.1 µM, appeared to block completely NK92MI cell-induced apoptosis of CHO-ICAM1 cells ( Fig. 4A ). When the concentration of caspase 3 inhibitor was below 0.1 µM, its blocking effect was reduced from −135 pm to −180 pm (the NK cells without a compound caused about −180 pm response). On the contrary, pretreatment with camptothecin, vinblastine, or BFA clearly enhanced NK92MI cell mediated apoptosis of CHO-ICAM1. However, the concentration required to show the initial enhancement was quite different among the three modulators: ~15 µM for camptothecin ( Fig. 4B ), ~0.4 µM for vinblastine ( Fig. 4C ), and ~0.2 µM for BFA ( Fig. 4D ).
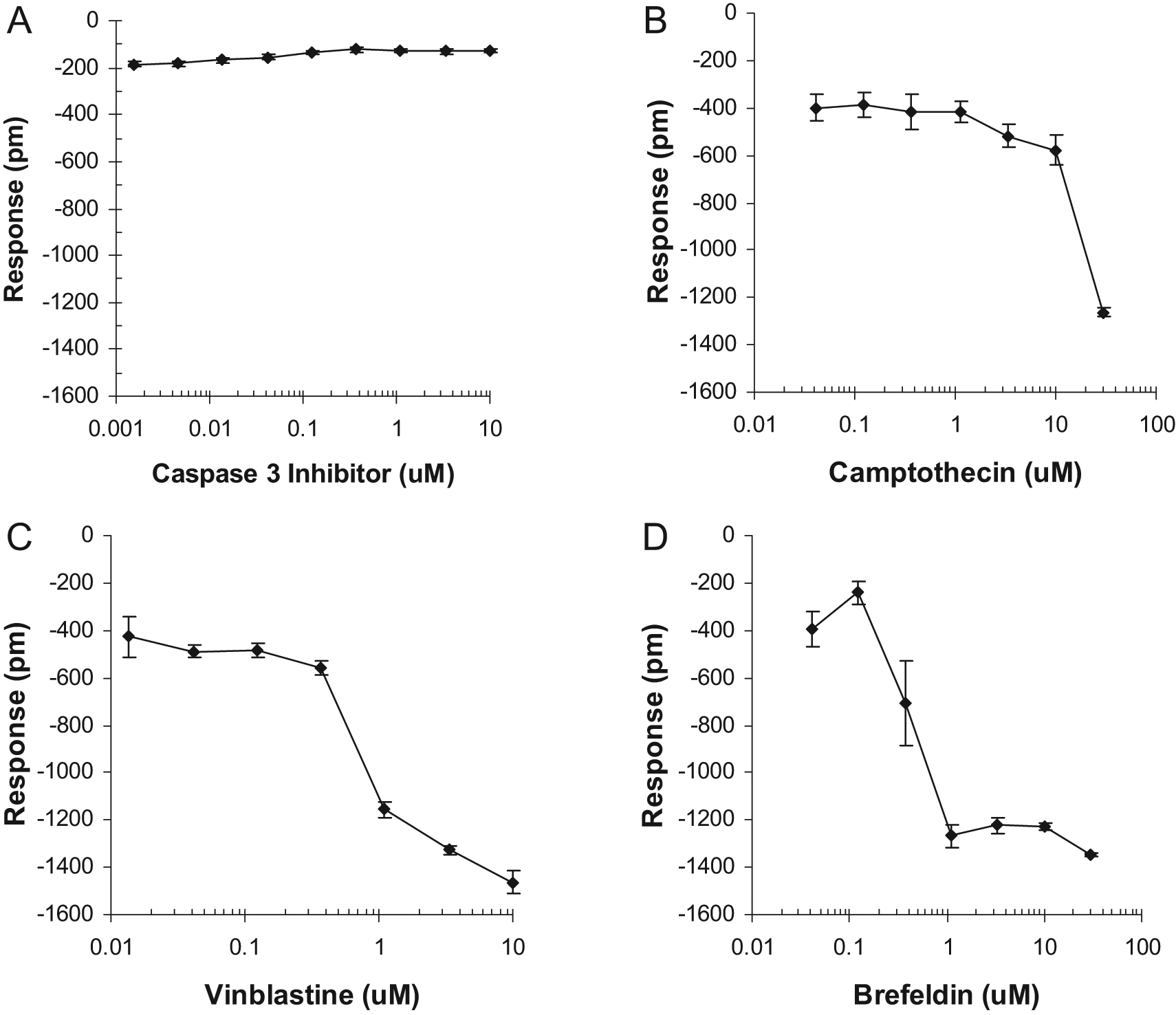
Effect of pharmacological reagents on NK92MI-mediated cytotoxicity of CHO-ICAM1 cells. (
The results help gain insights into the correlation between the cellular events and the optical signals. The caspase 3 inhibitor used in this study is known to be a potent cell-permeable and irreversible blocker of caspase 3 activity and also to have less inhibitory effect on the function of caspase 6, caspase 7, caspase 8, and caspase 9, all of which are important for apoptosis. Therefore, its ability to inhibit the normal CHO-ICAM1 cell response to NK92MI cells was indicative of the blockage of NK92MI-mediated apoptosis of CHO-ICAM1, suggesting that the apoptosis is mainly through caspase signaling pathways, as reported in the literature.20–22
Camptothecin blocks the cell cycle in S-phase at low dose and induces apoptosis at high dose in a large number of normal and tumor cell lines via cell cycle–dependent and –independent processes. It binds irreversibly to the DNA-topoisomerase I complex; therefore, it is inhibitory to DNA replication. 23 In our study, it had little effect on NK92MI-induced apoptosis at low concentrations (0.1–10 µM) but significantly promoted cell death at 30 µM, showing a steep change from approximately −400 pm to approximately −1200 pm. Interestingly, camptothecin treatment alone only had 23% of the effect relative to camptothecin plus NK92MI cells (data not shown).
Vinblastine is an anticancer drug. It binds to tubulin and inhibits microtubule formation, resulting in disruption of mitotic spindle assembly and arrest tumor cells in the M phase of the cell cycle.24,25 Based on the CHO-ICAM1 cell response curve, vinblastine is apparently more potent, requiring only 1 µM to show an initial effect. The change is also steep, from approximately −400 pm to approximately −1400 pm in a very small concentration range of 0.3 to 1 µM. With 10 µM, the response reaches a maximum of approximately add −1500 pm. Surprisingly, the CHO-ICAM1 cell response to vinblastine treatment alone was much smaller, only 28% relative to the treatment of vinblastine plus NK92MI cells (data not shown).
BFA is a fungal metabolite that specifically and reversibly blocks protein transport from the ER to the Golgi apparatus in many cell types, and prolonged exposure can induce apoptosis.26,27 In this study, it appeared to have the strongest effect, requiring only 0.2 µM to show a clear initial response. The effective concentration range was very narrow, from ~0.2 µM to 1 µM, triggering a sharp CHO-ICAM1 cell response from approximately −400 pm to approximately −1300 pm. The response was then maximized at higher concentrations of >1 µM. BFA treatment alone was clearly more toxic than the other modulators, but only 37% relative to the treatment of BFA plus NK92MI cells (data not shown).
To confirm the negative DMR signal indeed reflected effecter cell–mediated target cell apoptosis, we have examined cell attachment as it is well known that dead cells typically have a very poor attachment to a surface. After a 16-h assay and during the PBS wash process for live/dead cell staining, we observed that at the E/T ratio of 0, the target cell attachment was greater than 98%, whereas at E/T = 8, only ~5% of target cells were attached on the surface. At an E/T ratio of 0 plus 1 µM BFA, there were about 90% of cells still attached on the surface. However, at an E/T ratio of 0.5 plus 1 µM BFA, only ~2% target cells were attached on the surface ( Fig. 5 ). Clearly, the negative cell response resulted from dead cells’ leaving the surface (mass loss).
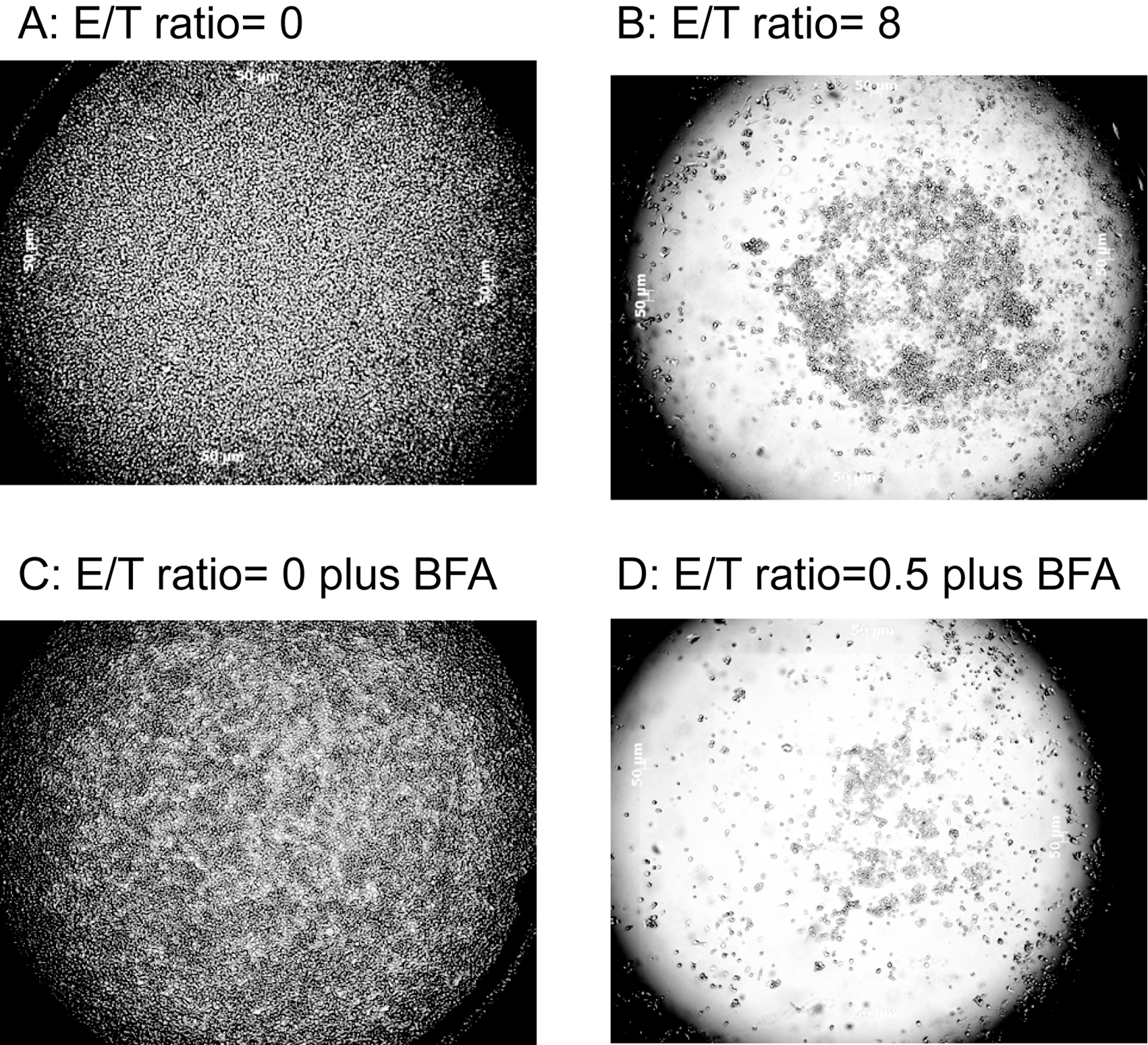
Microscopic images of CHO-ICAM1 cell confluency after Epic cell assay. (
Taken together, we have clearly observed distinct optical cell responses of the target CHO-ICAM1 cells to NK92MI cells in the absence or presence of four modulators known to act on the target cells through different mechanisms. As stated before, with NK92MI cells alone at the E/T ratio of 0.5, the maximum target CHO-ICAM1 cell response was approximately −180 pm. Except for caspase 3 inhibitor, all three other modulators had less effect when used alone but synergistically promoted target cell death when being treated along with the effecter NK92MI cells and had various levels of potency, resulting in a maximum effect ranging from approximately −1200 pm to approximately −1500 pm. In a separate study, we found that the DMR signal from a confluent monolayer of CHO-ICAM1 was ~1600 ± 204 pm. Accordingly, −1500 pm response could be translated to the detachment of the vast majority of the target cells from the sensor surface. It seemed that the optical sensor was more sensitive to the inhibition of overall cellular transportations (BFA) and less sensitive to the cell cycle arrest, even though it appeared to be able to distinguish whether the arrest was at the M phase (vinblastine) or S phase (camptothecin). In short, the data suggested that the Epic system may be useful for identifying novel chemicals that interfere with NK92MI cell–mediated cytotoxicity.
Effect of Modulators on Caspase Activity
The activity of caspases-2, -3, -6, -8, and -9 was further assessed with conventional enzymatic assays. Target CHO-ICAM1 cells pretreated with a modulator at a concentration (10 µM caspase 3 inhibitor, 30 µM camptothecin, 3 µM vinblastine, and 1 µM BFA) that clearly showed cytotoxic effect when coupled with NK92MI cells were lysed according to the protocol of EMD Biosciences. These cell lyses were used for the standard caspase assay with fluorescently labeled substrates in the absence or presence of an FMK-derived substrate. The results are summarized in Figure 6 . As expected, there was barely any detectable caspase activity in target cells alone (buffer control) and small activities with NK92MI cells without pretreatment of a compound (the RPMI medium control). Caspase 3 activity was very strong in the samples pretreated with the three toxic modulators (camptothecin, vinblastine, and BFA) but was abolished in the presence of the caspase 3 inhibitor. The enzymatic activity for both caspase 2 and 6 was high but negligible for caspase 8 and 9 in the samples pretreated with all four modulators. It indicated that the NK effect on the target cells is mainly through caspases 3, 2, and 6 but unlikely via caspases 8 and 9. The results further confirmed that the negative optical signals observed were most likely the reflection of target cell apoptosis.
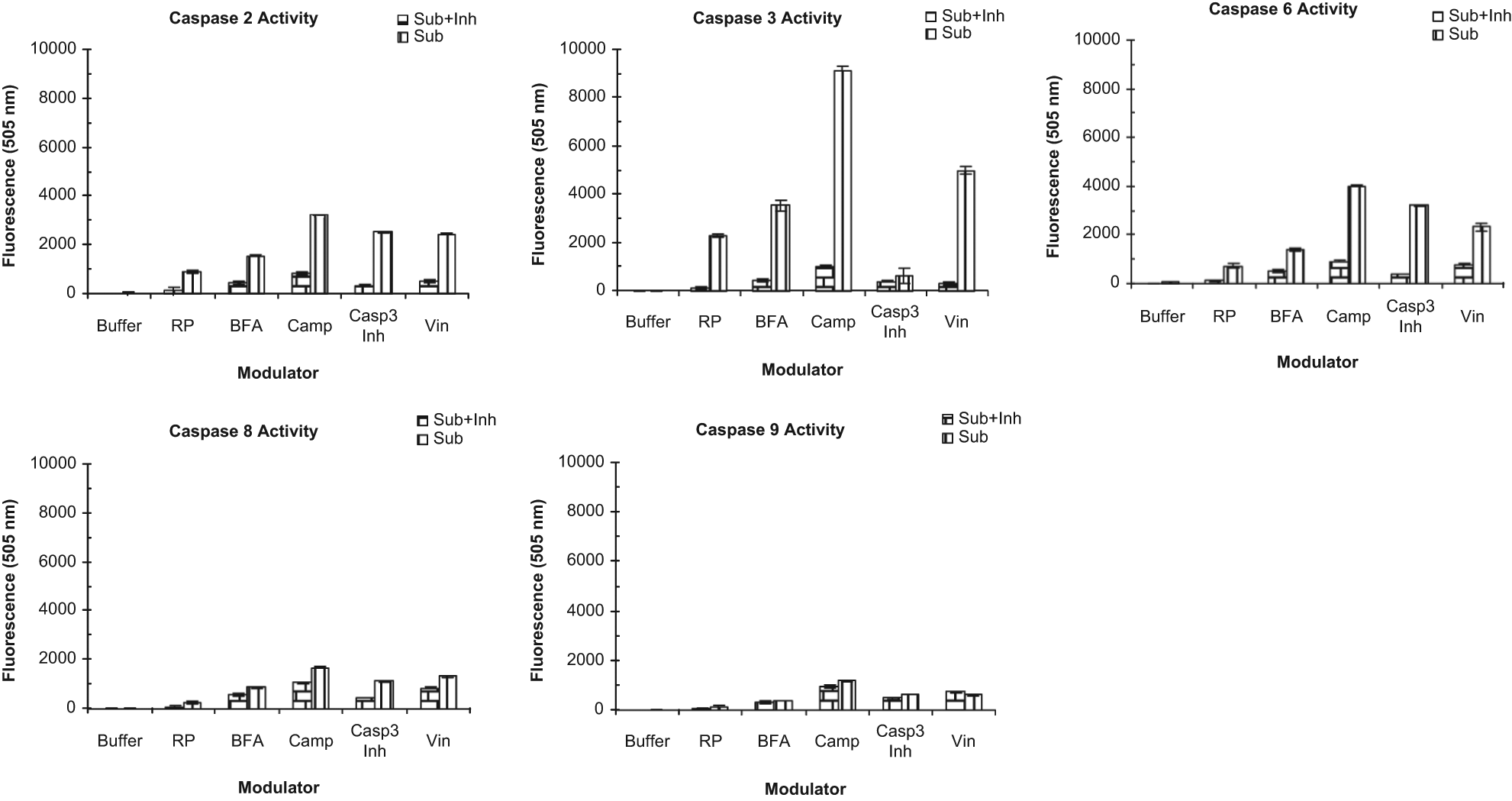
Conventional caspase enzymatic activity. CHO-ICAM1 cells were treated with a pharmacological reagent at a given concentration (see below), each in four replicates, for 1 h followed by incubation with NK91MI cells at the effecter cells to target cell™ (E/T) ratio of 0.5 for 16 h. The resulting cells were lysed and assayed for enzymatic activity of caspases 2, 3, 6, 8, and 9 according to the protocol provided by EMB Biosciences. Briefly, the cell lysate was incubated with a fluorescently labeled substrate in the presence or absence of its corresponding fluoromethyl keton (FMK)–derived substrate. The fluorescent signal resulting from the enzymatic activity of a caspase, defined by the substrates used in the reaction, was measured by Perkin Elmer VictorX4 reader at ~505 nm and shown in the y axis corresponding to the various treatments (x axis). The treatments were CHO-ICAM1 cells alone (buffer control), CHO-ICAM1 plus NK cells (RP), 1 µM brefedin A plus natural killer (NK) cells (BFA), 30 µM camptothecin plus NK cells (Camp), 10 µM caspase 3 inhibitor Z-DEVD-FMK plus NK cells (Casp3), and 3 µM vinblastine plus NK cells (Vin). Caspase substrates (Sub)/inhibits (Inh) Z-VDVAD-AFC (sub)/–FMK (inh) for caspase 2, Ac-DEVD-AFC (sub)/FMK (inh) for caspase-3, Ac-VEID-AFC (sub)/-FMK(inh) for caspase 6, Ac-IETD-AFC (sub)/-FMK (inh) for caspase 8, and Ac-LEHD-AFC (sub)/-FMK(inh) for caspase 9.
Conclusions
We have demonstrated a high-throughput label-free cell-based assay for the study of effecter cell–mediated target cell cytotoxicity with the Epic system. With initial tuning of the E/T ratio and interaction time, we believe that the assay may be generally applicable to a broad range of living cell-cell communication studies. Using the assay, the impact of effecter cells on target cells is directly monitored using optical sensors in a 384-well plate. Moreover, the assay is performed using whole live cells and measured in real time, which is more physiologically relevant to actual cellular conditions. With its sensitivity to distinguish different cellular events with optical signals (e.g., cell-cycle arrest vs inhibition of cellular transportation), new insights into the mechanism of various cell-cell communications may be obtained. Therefore, it presents a new powerful tool for assessing the effect of a molecule on a particular cell-cell interaction and discovering new drugs for the diseases involving such interaction.
Footnotes
Acknowledgements
We appreciated Dr. Norman Fontaine’s help with some of the early-stage study of cell-cell communication with an angular system. We would like to thank the Corning Epic team for their excellent support. We also thank Dr. van der Merwe for his pioneer work and great review on cell-cell interaction.
The authors declared internal potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received internal financial support for the research, authorship, and/or publication of this article.