
Abstract
Study Design:
Systematic review.
Objectives:
Cervical arthroplasty is an increasingly popular treatment of cervical radiculopathy and myelopathy. An understanding of the potential adverse events (AEs) is important to help both clinicians and patients. We sought to provide a comprehensive systematic review of the AEs reported in all randomized controlled trials (RCTs) of cervical disc arthroplasty in an attempt to characterize the quality of reporting.
Methods:
We conducted a systematic review of MEDLINE and Web of Science for RCTs of cervical disc arthroplasty reporting AEs. We reported the most frequently mentioned AEs, including dysphagia/dysphonia, vascular compromise, dural injury, and infections. We recorded the presence of industry funding and scored the quality of collection methods and reporting of AEs.
Results:
Of the 3734 identified articles, 29 articles met full inclusion criteria. The quality of AE reporting varied significantly between studies, and a combined meta-analysis was not feasible. The 29 articles covered separate 19 RCTs. Eight studies were US Food and Drug Administration (FDA) investigational device exemption (IDE) trials. Rates were recorded for the following AEs: dysphagia/dysphonia (range = 1.3% to 27.2%), vascular compromise (range = 1.1% to 2.4%), cervical wound infection (range = 1.2% to 22.5%), and cerebrospinal fluid leak (range = 0.8% to 7.1%).
Conclusions:
There is a lack of consistency in reporting of AEs among RCTs of cervical arthroplasty. FDA IDE trials scored better in AE event reporting compared to other studies. Standardized definitions for AEs and standardized data collection methodology are needed to improve future studies.
Introduction
Anterior cervical discectomy and fusion (ACDF) is a common procedure in the management of cervical degenerative disc disease. This procedure may lead to loss of motion at the operated level and was suspected to increase the incidence of adjacent level disc degeneration. 1,2 Recently, cervical disc arthroplasty (CDA) emerged as an alternative that would preserve motion at the index disc level. 3 –6 However, CDA has been associated with increased risk of unique complications such as heterotopic ossification, device migration, and segmental kyphosis. 3,4,7,8 Furthermore, multiple CDA disc types have been approved by the US Food and Drug Administration (FDA), with each disc type conferring unique advantages and disadvantages relative to ACDF and nonsurgical modalities. 6,8
Outcomes following ACDF and CDA have been compared across several prior studies. 7,9 –11 While both procedures share similar surgical approaches, an understanding of the unique risks of each procedure is important for patient safety and optimizing preoperative risk counseling. However, inconsistencies in adverse event (AE) reporting related to CDA have limited the interpretation of the clinical risks associated with the procedure. 12 Reporting of AEs in randomized controlled trials (RCTs) in general has been shown to be highly heterogeneous with significant variability. 13 Some authors have suggested that the presence of conflicts of interest has the potential to distort AE reporting among RCTs. 14 A clear understanding of how AEs are defined and reported in RCTs of CDA can help surgeons better interpret the results of trials comparing CDA to ACDF and thereby improve surgical decision making. The present study is a comprehensive systematic review of the quality of AE reporting in RCTs of CDA.
Methods
Study Search
We conducted MEDLINE and Web of Science database searches with the following search algorithm: “cervical” and (“arthroplasty” or “total disc replacement” or “artificial disc replacement” or “total disk replacement” or “artificial disk replacement”) and (“complications” or “outcomes” or “adverse events”). The search returned 3734 citations (Figure 1). The search period ended on May 9, 2017.
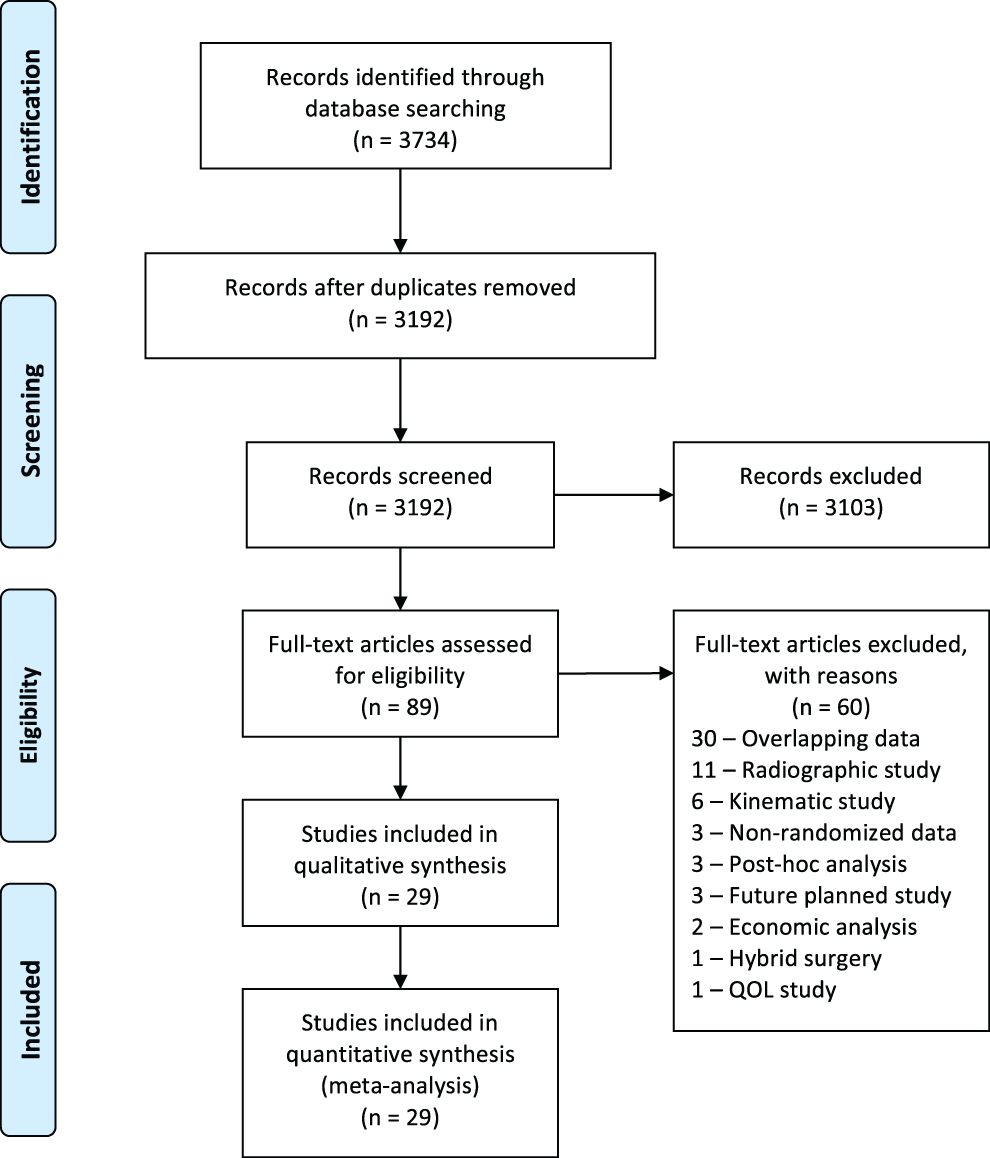
PRISMA flow diagram for selection of studies based on inclusion criteria during systematic review.
Inclusion and Exclusion Criteria
Only RCTs comparing CDA to anterior discectomy and fusion were included in this systematic review because of their superior evidence level compared to cohort studies. 15 We imposed no restrictions on publication status. Animal, in vitro, biomechanical, kinematic, and radiologic studies were excluded. Studies that only published the results of subgroup analyses of other RCTs were reviewed, and we excluded studies that reported redundant data from the same patient populations.
Data Collection
Two reviewers (JCX, CG) independently conducted data extraction from the 29 included articles. The extracted data sets were compared to confirm accuracy with a third reviewer (MS) arbitrating any disputes in abstracted data from different reviewers. The level of evidence of each of the included articles was assessed using the Oxford Centre for Evidence Based Medicine (OCEBM) Level of Evidence 2 classification system. 15
From the eligible articles, we obtained the following information: study type, publication year, sample size, number of operated levels, follow-up duration (months), average age of patient cohort, artificial disc type, definition of AEs, and number and type of AEs. We analyzed all AEs reported, regardless of the study’s definition of what counted as an AE. For studies that report both number of events and number of patients for AE, we only recorded the number of patients. If a study explicitly stated that no event occurred, the data was recorded appropriately.
We documented whether industry funding was received based on disclosures. Studies where corporate or industry funds were used in support of the work, or where authors received royalties, consultant fees, or research support from the producer of the investigational device were recorded as having industry funding.
To assess the risk of bias for each study, 2 reviewers independently investigated the individual studies (JCX, CG) and used The Cochrane Collaboration’s tool for assessing risk of bias. 16 Bias risk assessment was performed at the study level. Inconsistencies in bias risk assessment were reconciled through discussion between 3 reviewers (JCX, CG, and MS).
Analysis
We assigned overall scores for the quality of collecting and reporting AEs for each included study. Scoring criteria were based on previously published scoring systems. 17,18 Scores for AE collection quality were based on whether collection, definitions, severity grading, timing, and methods for statistical analysis of AEs were reported in the article. Studies must have specifically mentioned AEs in their statistical methods to have a positive score.
AE results reporting scores were based on whether AEs were reported, categorized, timed, statistically analyzed, and whether reoperations were included. Both AE collection quality and AE results reporting scores received 1 point for each present category for a maximum score of 5. To assess differences in scoring, a 2-sample Student’s t test was performed using Microsoft Excel’s TTEST function with 2 tails assuming unequal variance. Statistical significance was set at an α level of .05. Due to inconsistent reporting and definitions of AE, a meta-analysis of this data was not possible.
Results
Study Selection
The initial 3734 retrieved citations were reviewed. After removing 542 duplicates, the titles and abstracts of 3193 publications were screened. After excluding 3103 citations, the full text was assessed in the remaining 89 articles for eligibility criteria. Full-text assessment resulted in 29 eligible articles included in the final analysis.
Study Characteristics
As shown in Table 1, the 29 included studies ranged in publication year from 2004 to 2017 and included 19 separate RCTs. Cohorts ranged in size from 10 to 276 patients. Follow-up time ranged from 12 to 84 months, with a mean of 40.7 months between all studies. The included studies were of the Mobi-C, ProDisc-C, Porous Coated Motion (PCM), Prestige LP, Prestige ST, Bryan, Kineflex C, and Discover disc types. 19 –26 Eight of these RCTs were FDA investigational device exemption (IDE) trials, which establish safety and efficacy in the process for FDA approval. Among the major FDA IDE trials, long-term data wasavailable up to 7 years for the ProDisc-C, PCM, Prestige LP, and Prestige ST devices, and up to 5 years in the Mobi-C 1- and 2-level trials. 19,22,27 –30 The ProDisc-C trial demonstrates that AEs continue to increase with time. The rates at 2- and 5-year follow-up of the ProDisc-C were 2.9% and 11.7%, respectively. 31,32 The 7-year data for the same trial reported a cumulative AE rate of 27%. 22
Summary of Included RCT Studiesa.
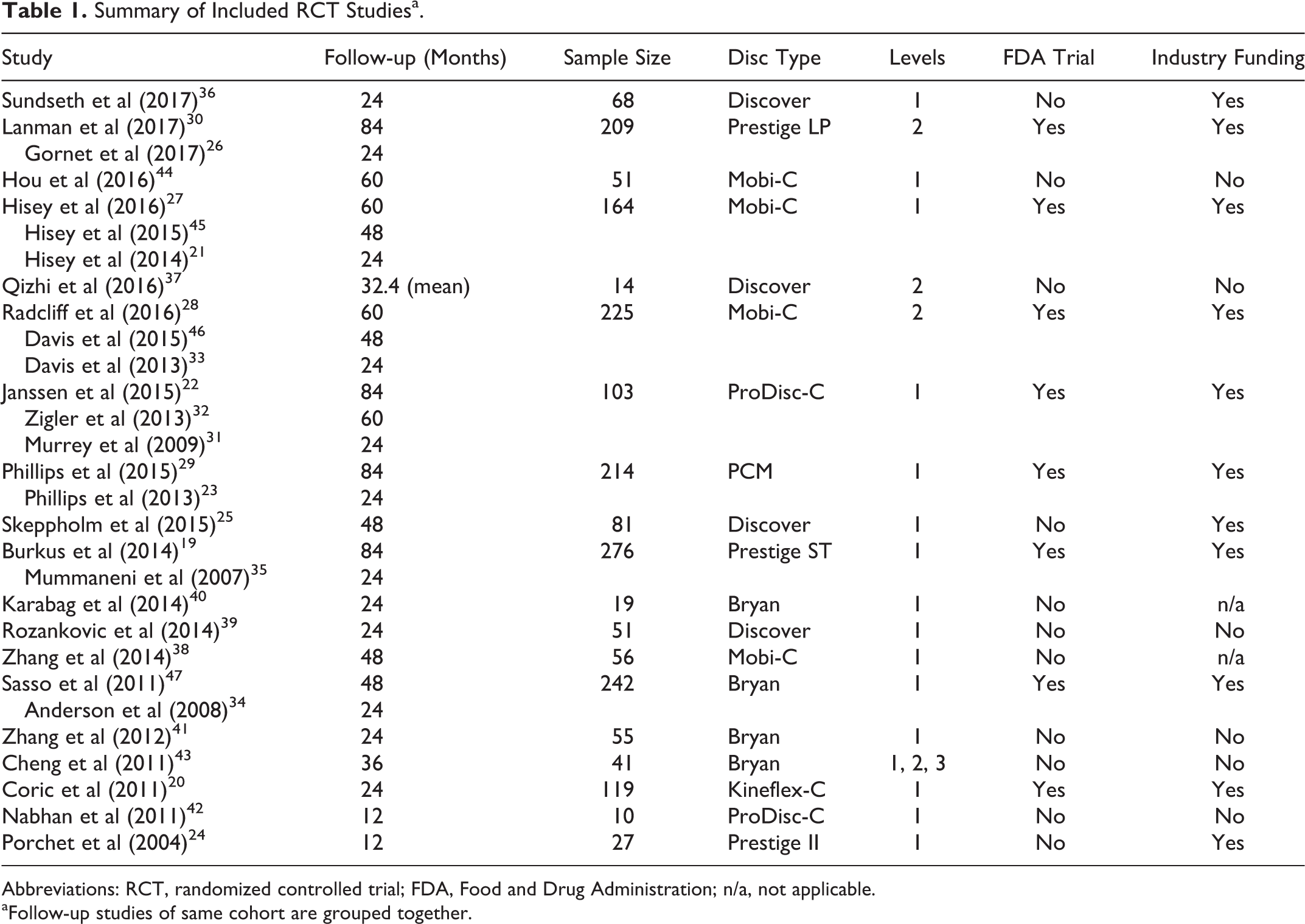
Abbreviations: RCT, randomized controlled trial; FDA, Food and Drug Administration; n/a, not applicable.
aFollow-up studies of same cohort are grouped together.
Industry funding was reported in all 8 FDA IDE trials, as well as 3 international studies. 20,21,23 –26,31,33 –36 Eight separate RCTs, all completed internationally, reported no industry funding or did not make any disclosures. 37 –44 As part of the FDA evaluation, AE reporting is required, whether the AEs appear related to the procedure or not. Nearly all (25 out of 29) of the reviewed articles reported at least one AE, but there was a lack of standardization regarding AE definitions, collection methods, or categorization. Average total scores for AE collection quality and AE results reporting for all studies were 2.34 and 2.59, respectively (Tables 2 and 3).
Overall Score of the Quality of Adverse Event Acquisition Methodology and Reportinga.
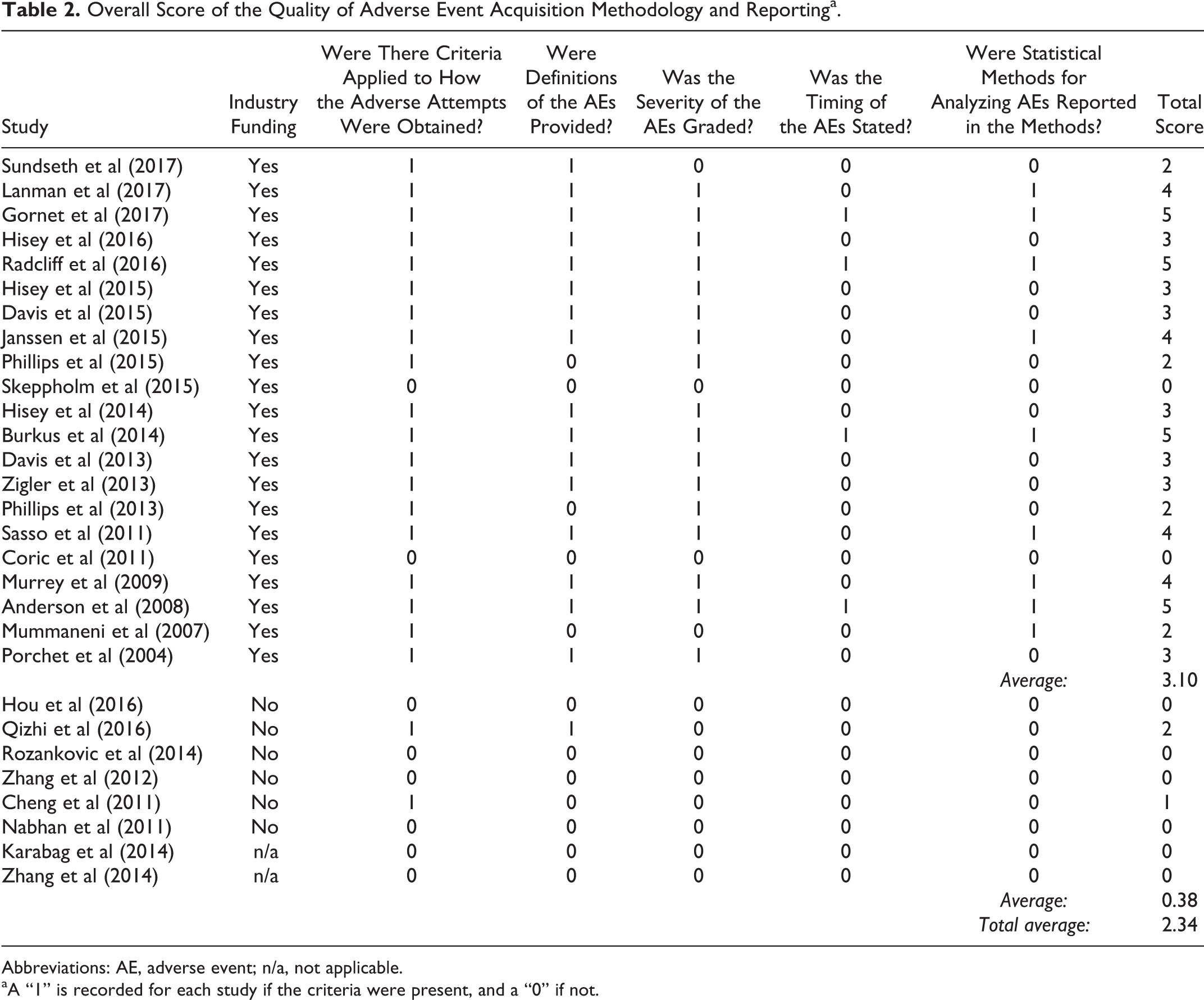
Abbreviations: AE, adverse event; n/a, not applicable.
aA “1” is recorded for each study if the criteria were present, and a “0” if not.
Overall Score of AE Results Reportinga.
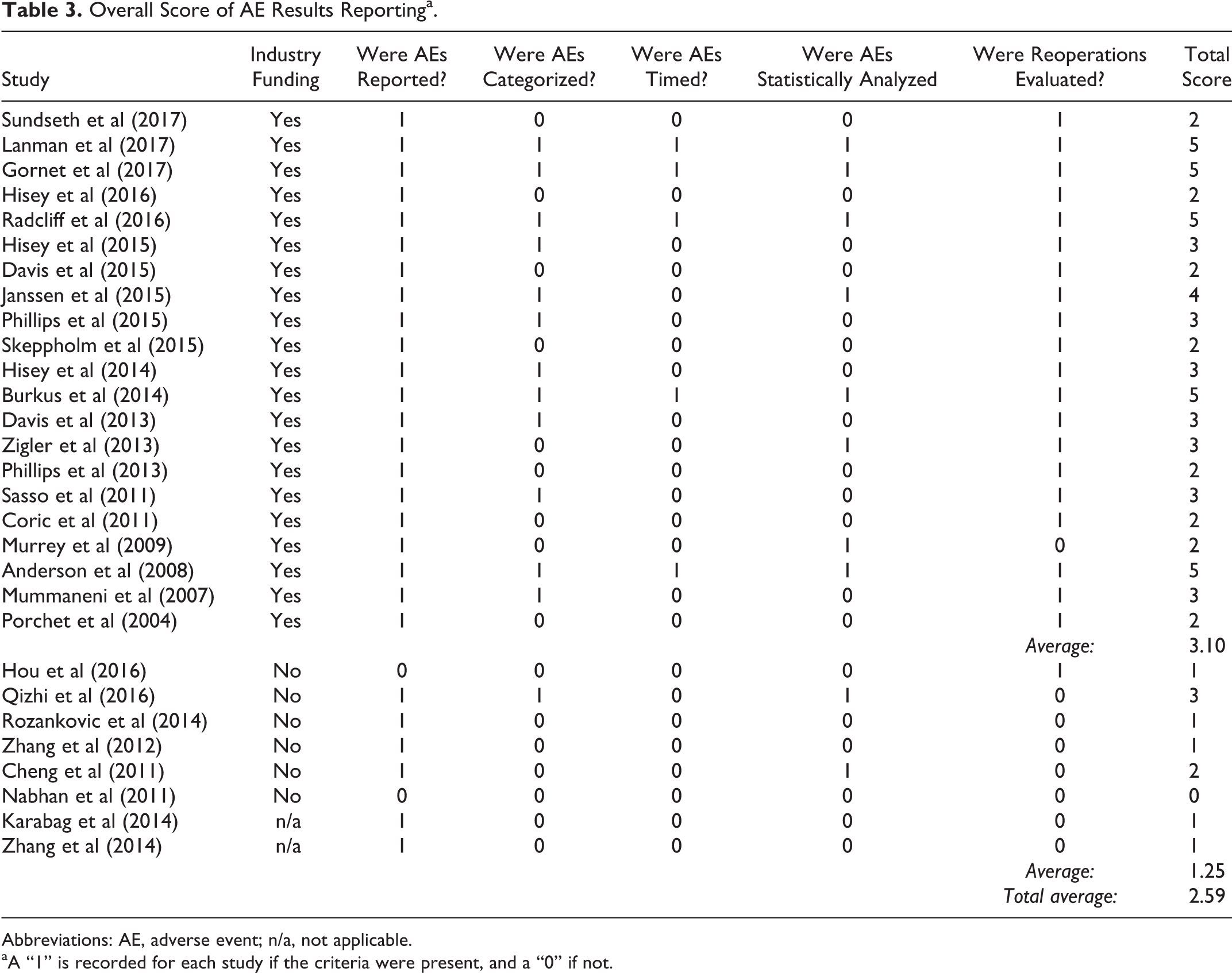
Abbreviations: AE, adverse event; n/a, not applicable.
aA “1” is recorded for each study if the criteria were present, and a “0” if not.
Bias risk assessment of the included studies demonstrated a discernible difference between RCTs with industry funding and those without (Table 4). Only one of the studies without industry funding described how participants, personnel, and outcome assessors were blinded, compared to 16 out of 21 studies that did have industry funding.
Bias Risk Assessment of 29 included RCTs.
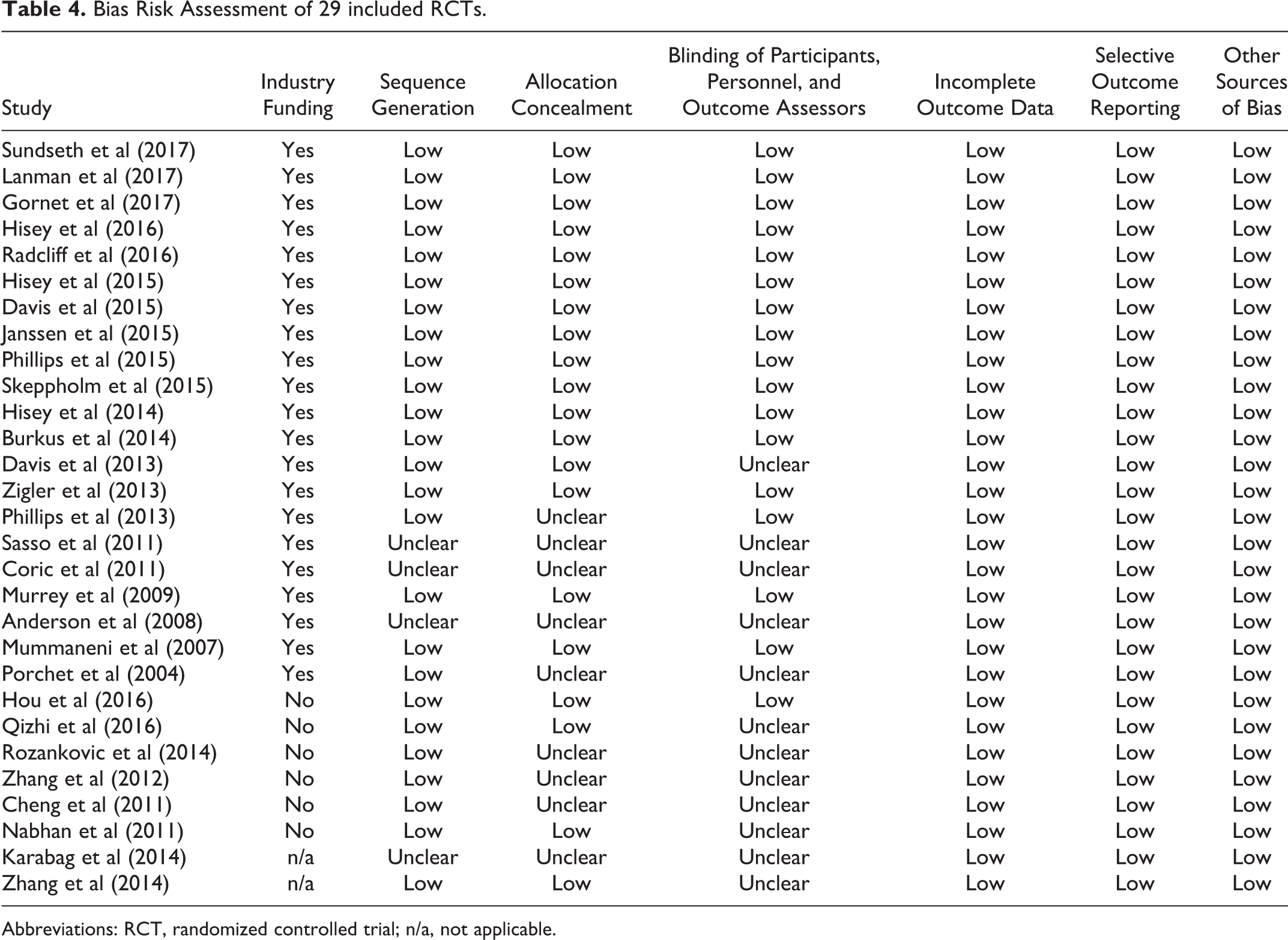
Abbreviations: RCT, randomized controlled trial; n/a, not applicable.
Adverse Event Definitions
We found significant heterogeneity across studies in the methodology for reporting and definition of AEs (Table 5). For example, Murrey et al 31 only reported AEs that were classified as “severe or life-threatening,” while other studies defined and reported AEs regardless of severity. 19,31,34 Qizhi et al 37 only reported complications that were related to the intervention, while all 8 FDA studies collected AEs regardless of causality. 19 –21,23,26,31,33,34 Individual AEs also varied in how they were assessed. A variety of different assessment tools were used to assess dysphagia: Phillips et al 23 used a Visual Analogue Scale (VAS); Skeppholm et al 25 and Sundseth et al 36 used the Dysphagia Short Questionnaire; Hisey et al 21 used the Functional Outcome Swallowing Scale; and Qizhi et al 37 used the Swallowing Quality of Life Questionnaire.
Individual Study Definitions of AEsa.

Abbreviations: AE, adverse event; WHO, World Health Organization.
aStudies that did not provide a definition for AEs are excluded from the table.
The Mobi-C (1- and 2-level) and Prestige LP FDA trials reported that independent committees of 3 members assessed complications in a blinded manner, and classified complications by severity and the likelihood that an event was related to the intervention. 21,26 –28,33,45,46 Porchet et al 24 assessed the severity of AEs according to the World Health Organization (WHO) recommendations, graded from 1 to 3. Authors of the Bryan disc and Prestige LP FDA trials categorized AEs using a modified WHO criteria, graded from 1 to 4. 26,30,34,47 The description of each grading criteria are presented in Table 5.
Overall AE Reporting
There was significant variation in the quality of AE reporting. Out of the 19 RCTs, 10 reported a total percentage of AEs, while the other 9 reported specific AEs or none at all. The Bryan, Prestige ST, Prestige LP, and Mobi-C trials included many nonsurgical AEs including cancer, gastrointestinal, cardiovascular, urogenital, and death. 19,21,22,26,33,34 Anderson et al 34 also included AEs related to anesthesia, and technical AEs including drill failure and surgical malpositioning. The Mobi-C, PCM, and Bryan disc trials only reported total AEs if they were deemed “serious” or “major,” and these are noted in Table 6. 23,29,33,34,45,47 In this review, when reports provided rates for both total AEs and serious AEs, only the latter were recorded.
Total Reported AEsa.
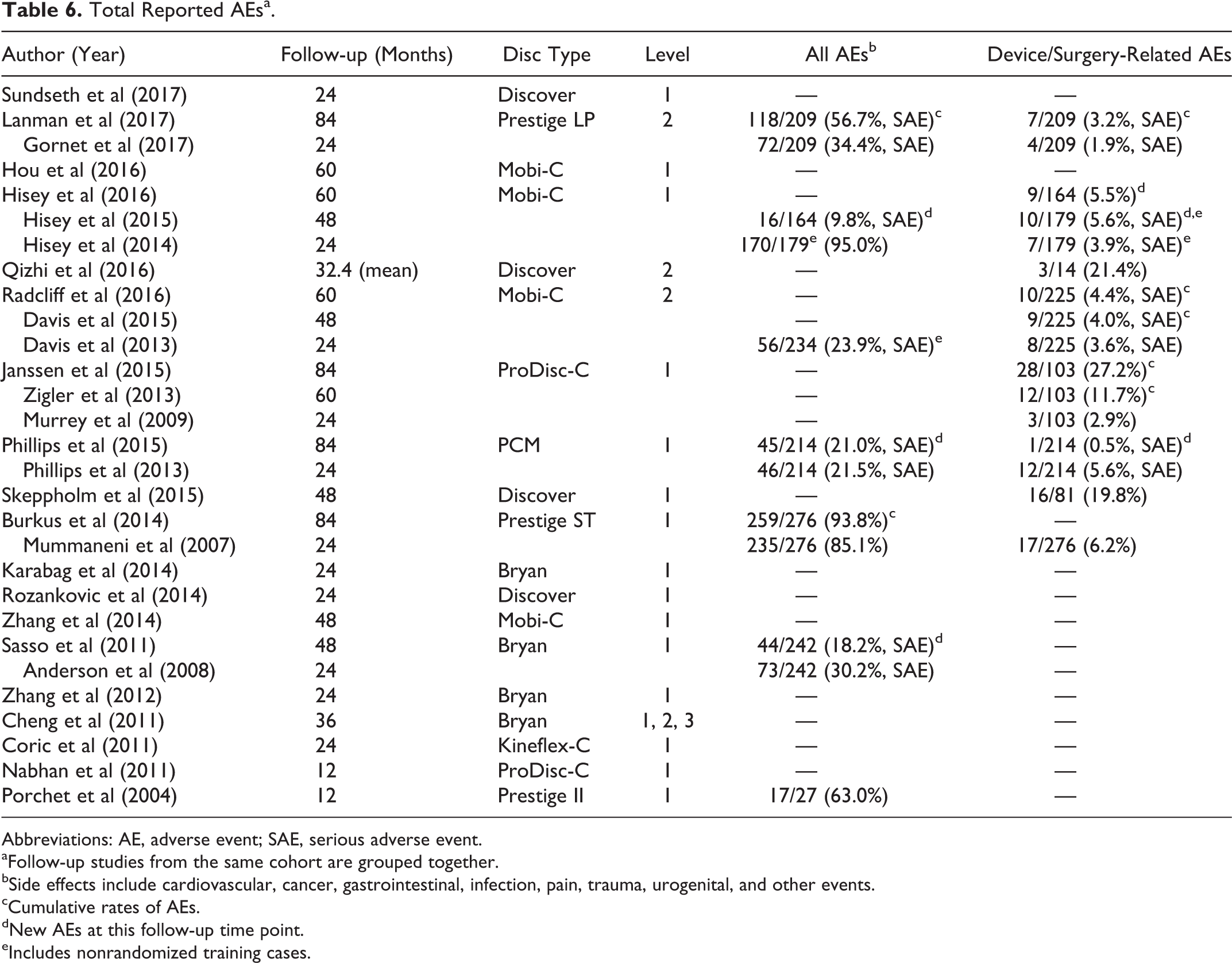
Abbreviations: AE, adverse event; SAE, serious adverse event.
aFollow-up studies from the same cohort are grouped together.
bSide effects include cardiovascular, cancer, gastrointestinal, infection, pain, trauma, urogenital, and other events.
cCumulative rates of AEs.
dNew AEs at this follow-up time point.
eIncludes nonrandomized training cases.
Common Postoperative AEs
Table 7 presents the most commonly reported AEs among the included studies. The most commonly reported AEs were dysphagia/dysphonia (range = 1.3% 30 to 27.2% 41 ), vascular compromise (range = 1.1% 21 to 2.4% 26 ), dural injury (range = 0.0% 21 to 7.1% 37 ), and cervical wound infection (1.2% 25 to 22.5% 19 ). Dural injuries included intraoperative durotomy and postoperative cerebrospinal fluid leaks. 34,37 Vascular compromise included intraoperative bleeding and/or postoperative hematoma formation. Gornet et al 26 provided an AE category of “vascular” but did not specify what this included or whether it was related to the operation. Burkus et al, 19 Mummaneni et al, 35 and Gornet et al 26 listed a general category of unspecified infections, which may have included infections unrelated to the surgical intervention. 19,35 Hisey et al 21 also reported a general category of infections that was subdivided into superficial cervical (3.4%), deep cervical (0.0%), other wound (0.6%), systemic (4.5%), and local (11.2%) infections. In our review, only cervical wound infections were included when specified, and other types of infections were excluded. Due to the heterogeneity of reported rates due to varying definitions and time points, an average rate of each individual AE could not be reasonably estimated.
Common Postoperative Complicationsa.
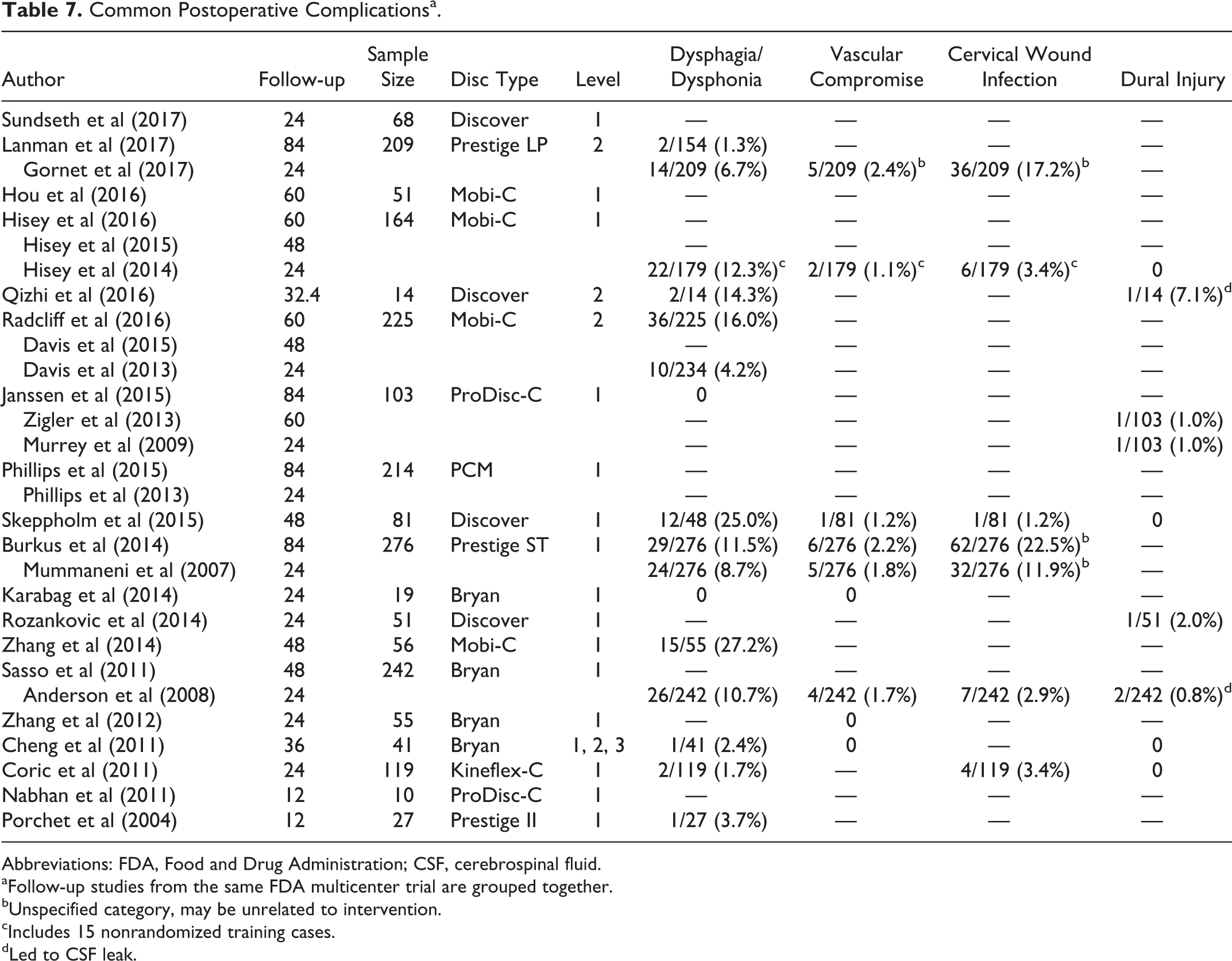
Abbreviations: FDA, Food and Drug Administration; CSF, cerebrospinal fluid.
aFollow-up studies from the same FDA multicenter trial are grouped together.
bUnspecified category, may be unrelated to intervention.
cIncludes 15 nonrandomized training cases.
dLed to CSF leak.
Issues with AE Methodology and Reporting Quality
Four of the included studies reported the timing of AEs. 19,26,28,34 Gornet et al 26 provided specific AE rates at the following time points: Operatively, 1 day to 4 weeks postoperation, 1.5, 3, 6, 12, and 24 months postoperation. Anderson et al 34 provided rates within 6 weeks of the operation, and between 6 weeks and 3 years postoperation. Burkus et al 19 provided rates of individual AEs at 24 and 84 months postoperation. Radcliffe et al 28 provided total AE rates at 6, 12, 18, 24, 36, 48, and 60 months postoperation, but did not include a breakdown of specific AEs. As previously mentioned, the Mobi-C and Prestige LP FDA trials assigned an independent committee to categorize and grade AE severity. 21,26 –28,33,45,46 All other remaining studies did not report how they graded AE severity. 19,22,25,31,32,37 –43
Quality of AE in Reports With and Without Industry Funding
Out of the 19 included RCTs, 11 reported industry funding, 20,21,23 –26,31,33 –36 while 6 studies reported no industry funding, 37,39,41 –44 and 2 did not provide disclosures. 38,40 Out of the 11 studies with industry funding, 8 were FDA IDE trials. 20,21,23,26,31,33 –35 Studies reporting no industry funding and those that did not provide disclosures were all completed outside the United States. The studies without industry funding scored lower compared to studies with industry funding in both average AE methods clarity (0.38 vs 3.10, P < .001) and results reporting (1.25 vs 3.10, P < .001). The studies with industry funding more consistently provided AE definitions, grading, timing, and data on subsequent surgeries compared to studies without any industry funding.
The Prestige LP FDA trial, Bryan FDA trial, and Prestige II trial with standardize AE categorization via the WHO severity grading. 24,26,30,34,47 These articles scored an average of 4.2 on methods clarity, significantly higher than the average of 2.8 in industry funded studies that did not use WHO grading (P < .05). The same articles scored 3.8 on results reporting, which was not significantly different from the average of 2.9 in industry funded papers without WHO grading. The Mobi-C trials (1- and 2-level) and Prestige LP FDA trials that used an independent committee to review AEs had average scores of 3.63 and 3.38 for methods clarity and results reporting, respectively. 21,26 –28,30,33,45,46 Studies that did not specifically report an independent committee scored 2.8 and 2.9, respectively, in the same categories. There was no significant difference between scores for studies that used an independent committee and those that did not.
Sundseth et al, 36 Skeppholm et al, 25 and Porchet et al 24 were non-FDA trials that reported industry funding. These studies averaged 1.67 and 2.00 in methods clarity and results reporting, compared to 3.33 and 3.30 for the same categories in the FDA trials (no significant difference).
Discussion
This study presents a comprehensive systematic literature review of AE methods and reporting quality associated with cervical arthroplasty and compares studies with and without industry funding. We sought to clarify potential AEs specific to cervical arthroplasty, to critically assess the quality of AE methodology and reporting among RCTs, and to identify practices that lead to useful AE results.
Adverse Events
Only 8 out of the 19 included RCTs we assessed provided a clear definition of AEs. The widely varying rates of reported AE across studies may reflect inconsistencies in how AEs are defined and reported. Burkus et al 19 and Hisey et al, 21 both FDA IDE trials, reported that more than 90% of their patients suffered from at least one AE, with no classification of severity. In contrast, Phillips et al, 29 another FDA IDE trial, reported that 21% of their patients suffered from at least one AE.
Postsurgical AEs develop as a result of myriad potential etiologies. Davis et al 33 and Phillips et al 23 both noted that the majority of the events reported were unrelated to the intervention. Coupled with the inclusion of AEs unrelated to the disc device or surgical intervention in all 6 of the 16 RCTs, the heterogeneity of etiologies underlying the incidence of postsurgical AE make it difficult to draw clinically relevant conclusions from the reported AE event rates. On the other hand, Mummaneni et al 35 reported only AEs in the perioperative period, which did not have a clear time frame. This may lead to underreporting of AEs that occur several months post-operation.
Upadhyaya et al 48 recently published a review of results from the ProDisc-C, Bryan, and Prestige ST FDA IDE trials and determined the reporting of AEs to be too heterogeneous to perform a combined analysis. 12 In their discussion, the authors note that the same AE could be categorized differently among various trials, further complicating the ability to compare across studies. We sought to summarize the most frequently reported AEs across all RCTs included in this study, which included dysphagia, vascular injury, dural injury, and infection. When pain was reported as an AE, it was the most common type of event among the included studies, although it was only categorized as an AE in 4 of the included studies. 19,21,22,49
Complications following CDA also include radiographic changes, such as heterotopic ossification and adjacent segment degeneration. 50 –54 More severe AEs, such as device migration or vertebral body fracture, may lead to subsequent surgeries. 55 These additional complications were not reported under AEs in each RCT, and were therefore not discussed in the present review. 3,56
Methods Clarity and Results Quality
Quantifying the risk for AEs following CDA is essential to both patients and surgeons during the informed consent process. Heterogeneity in the quality and clarity of AE reporting has been documented in other spinal surgery cohorts. Hiratzka et al 17 reviewed AEs reported by RCTs investigating lumbar fusion and found a high degree of heterogeneity in both the quality and consistency of reporting AEs. In the present study, we used the same scoring system as Hiratzka et al 17 and Anderson et al 18 to evaluate the included RCTs, ultimately yielding similar results.
Eight out of 11 studies with industry funding were FDA-regulated trials. As such, it was unsurprising that RCTs with industry funding had higher average methodological clarity and reporting results scores relative to RCTs that did not have industry funding. While there is always a potential bias that stems from conflicts of interest created by the financial relationship between study sponsors and investigators, most studies reported measures to minimize this potential through unrestricted grants and/or independent reviewers. We found that industry-funded studies were more likely to provide definitions and grade the severity of AEs relative to studies that did not report industry funding.
We found that the industry-funded studies that were not FDA trials scored lower on methods clarity and results reporting compared with the FDA trials. This provides support for the idea that the higher quality of AE reporting is associated more with the presence of FDA regulation than with the presence of industry funding. This suggests that surgeons should remain cautious when evaluating the results of non–FDA-regulated clinical trials, as these studies may underreport AE incidence rate.
Standardized systems to record, assess, and report AEs have been proposed by several authors. 17,57,58 Street et al 58 developed and validated a Spine Adverse Event Severity (SAVES) system, a simple questionnaire about perioperative complications. A Consolidated Standards of Reporting Trials (CONSORT) publication also provides guidelines for AE reporting, including clear definitions, severity grading, separation, and categorization of AE. 57 Among the included studies, studies that used these standardized reporting systems and approaches had higher reported rates of AEs. 58,59 The SAVES, CONSORT, or other guideline should be used in future studies comparing ACDF to CDA to better organize and standardize reporting of AEs. Ultimately, improving the quality of AE reporting across trials can improve preoperative risk counseling and surgical decision making by better informing patients and surgeons of the risks associated with CDA relative to other treatment modalities.
Recommendations
Future RCTs that report on AEs should strive to provide relevant data to help us understand the risks of a new device. The scoring system for AE methods clarity and results reporting from Hiratzka et al 17 and Anderson et al 18 provides a basis for 3 important principles to assess AEs. First, investigators should choose an established definition for general categories of AEs. Second, the severity of an AE should be graded. The WHO criteria for AEs has been applied to several studies included in this study. Third, studies should document the timing of when individual AEs were discovered. As evident by studies with longer follow-up, AEs continue to be reported up to 7 years postoperation.
Limitations
Many studies included in our screening process reported data from overlapping RCT cohorts. We attempted to limit redundant data in our systematic review by only including unique data from each RCT. As the definitions of AEs varied widely, we were limited in our ability to combine results for analysis. The requirements of FDA studies to report all AEs may have led to an overestimation of the actual incidence of AEs related to CDA, as even those AEs that were not associated with the procedure were reported in these studies. In addition, the number of studies with no industry funding was small, and all were completed internationally. This prevents an effective comparison between studies with and without industry funding.
Conclusions
Significant heterogeneity exists in the reported rates of AEs following CDA. This heterogeneity is likely due to the substantial variation in methodology for collecting and definitions for reporting AEs rates across studies. Studies that were FDA trials, regulated by stringent reporting guidelines, were associated with significantly higher scores for both methods clarity and reporting quality of AE. However, the variation in definitions and categorization undermine the ability to compare and combine results. Studies that were unregulated were less likely to provide clear definitions and reported a limited amount of AE. More consistent categorization and severity grading of AEs are necessary in order to compare results across studies and to provide meaningful clinical data.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.