
Abstract
Introduction: Acute respiratory response syndrome (ARDS) leads to increased permeability of the endothelial-epithelial barrier, which in turn promotes edema formation and hypoxemic respiratory failure. Although activated neutrophils are thought to play a significant role in mediating ARDS, at present the contribution of neutrophil extracellular traps (NETs) to lung endothelial barrier function is unclear. Methods: To clarify their role, we co-cultured in vitro NETs induced by phorbol myristate acetate (PMA)–activated neutrophils with lung endothelial cell monolayers and examined the barrier function of lung endothelial cells by immunofluorescence microscopy and albumin permeability in a double-chamber culture method. Results: Co-culture with stimulated neutrophils increased the albumin permeability of the human pulmonary artery endothelial cell (HPAEC) monolayer and altered cytoskeleton F-actin and vascular endothelial-cadherin in cell-cell junctions. Hyperpermeability to albumin and histological alterations were prevented by inhibition of NET formation with peptidyl arginine deiminase inhibitor or a neutrophil elastase inhibitor and were also prevented by increased degradation of NET structure with DNase. Conclusion: This in vitro experiment shows that altered HPAEC barrier function and increased albumin permeability are caused by the direct effect of PMA-induced NETs and their components. NET formation may be involved in the increased vascular permeability of the lung, which is a common feature in ARDS of various etiologies. These insights may help generate novel approaches for medical interventions.
Keywords
Introduction
Acute respiratory distress syndrome (ARDS) is an acute inflammatory lung injury characterized by hypoxemic respiratory failure as a consequence of increased permeability of the endothelial-epithelial barrier, alveolar damage, and pulmonary edema. 1 The pathogenesis of ARDS is complex, and the syndrome has a high mortality rate in critically ill patients. 2 Despite significant advances in mechanical ventilation aimed to better protect the lungs, ARDS remains difficult to prevent, reduce, or treat effectively. 3
Recent research has identified a novel antibacterial strategy: neutrophil extracellular traps (NETs), which localize to and eliminate pathogens. 4 These NETs are characterized by chromatin decorated with cytosolic and granular proteins. 4 They immobilize or trap various pathogens, thus preventing their dissemination. 5 However, similar to the excess production of inflammatory mediators, the excessive presence of NETs—and in particular NET-bound components—also has harmful effects. 6 For instance, NETs contain histone, neutrophil elastase (NE), cathepsin G, and myeloperoxidase (MPO), all of which are cytotoxic to endothelial cells.7–9
Neutrophil extracellular traps are found not only at sites of infection and acute inflammation but also in the bloodstream, where they are known as circulating cell-free NETs. 10 Researchers have hypothesized that circulating cell-free NETs interact with platelets, leucocytes, and the vascular endothelium in the lung to induce ARDS.11–13 In patients with ARDS11,14–16 and in murine models of lung injury, 14 NETs develop in response to a variety of infectious stimuli and contribute to the injury.
Direct evidence of lung hyperpermeability caused by NETs and circulating cell-free NETs is still lacking. Therefore, to examine whether NET formation is involved in lung hyperpermeability during acute inflammation, we co-cultured human pulmonary endothelial cell monolayers with neutrophils after pretreatment with phorbol myristate acetate (PMA). We show that the formation and components of NETs affect lung endothelial barrier function and increase endothelial permeability. Inhibition of NET formation prevents cytoskeleton remodeling and alterations of vascular endothelial (VE)-cadherin in the cell-cell junction.
Materials and methods
Isolation of neutrophils
A lithium heparin blood collection tube (BD Vacutainer; Becton, Dickinson and Company, Franklin Lakes, NJ, USA) was used to collect peripheral blood by venipuncture from 10 healthy Japanese adult volunteers (5 men and 5 women) with a median age of 30 years (range, 18–54 years). Volunteers were recruited at Aichi Medical University by advertisement. Exclusion criteria were a negative history of malignant, degenerative, or transmitted disease; type 2 diabetes; and use of corticosteroids or other immunosuppressive agents at the time of the study. Blood collection was performed with the understanding and consent of each participant. Polymorphonuclear neutrophils (PMNs) were isolated by discontinuous density gradient centrifugation on 1-Step Polymorphs (Accurate Chemical and Scientific Corporation, Westbury, NY). To prevent neutrophil activation during the separation procedure, we resuspended pellets slowly and kept the vortex setting in the low- to mid-range so that cells were not activated. Freshly isolated PMNs were resuspended with phenol red-free RPMI-1640 medium containing 2 mM L-glutamine (Wako Pure Chemicals, Osaka, Japan) supplemented with 2% heat-inactivated fetal bovine serum (FBS). This method yields samples with viability greater than 95% as assessed by trypan blue dye exclusion. To be sure that PMNs were not activated during isolation, we performed flow cytometry analysis to show the absence of CD11b, an antigen on the cell surface of neutrophils whose expression increases considerably when neutrophils are activated. Cell density was determined by a manual-counting method in which a user counted cells in microscope fields with a hemocytometer.
All experiments were approved by the Institutional Review Board of Aichi Medical University (17-H341) and were carried out in accordance with the Declaration of Helsinki. Written informed consent was obtained from each participant.
Endothelial cell culture
Human pulmonary artery endothelial cells (HPAECs) were purchased from Lonza (Basel, Switzerland) and cultured on collagen 1-coated culture flasks in EGM-2 medium (Lonza, CC-3156) containing an EGM-2 SingleQuots kit (Lonza, CC-4176). This growth supplement includes human epidermal growth factor, vascular endothelial growth factor, R3 insulin-like growth factor-1, ascorbic acid, hydrocortisone, human fibroblast growth factor-beta, and gentamicin/amphotericin-B. Culture media were refreshed every other day. After reaching confluence (approximately 6 days), cells were detached by trypsinization using 0.25w/v% trypsin containing 1 mM EDTA (Wako Pure Chemicals, Osaka, Japan) and split 1:3 for renewed passages. We used cells that reached the third passage.
Detection of neutrophil extracellular traps by immunolabeling
Freshly isolated neutrophils (5 × 105–106 cells/mL) were seeded onto 0.001% poly-D-lysine–coated glass coverslips, allowed to adhere, and stimulated with 25 nM PMA for 2 h at 37°C in 5% CO2/95% air. Then, the cells were washed 3 times with phosphate-buffered saline (PBS) to remove PMA by flicking the coverslips. After the third wash, the cells were resuspended with fresh phenol red-free RPMI-1640 medium containing 2 mM L-glutamine supplemented with 2% heat-inactivated FBS and further incubated for 4 h. The cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and blocked with 1% skim milk. 17 Cells were subsequently incubated overnight with diluted primary antibodies (monoclonal anti-NE antibody [0.7 μg] and anti-MPO antibody [0.1 μg]) and diluted secondary antibodies (Alexa Fluor-647-conjugated antibody [2 μg] and Alexa Fluor-488-conjugated antibody [2 μg]; all antibodies from Abcam, Cambridge, UK). After staining DNA with 1 μg/mL of Hoechst 33342 for 5 min, specimens were mounted in Mowiol (Sigma-Aldrich), and NETs were visualized with a Zeiss confocal laser scanning microscope (CLSM) 710 system (Goettingen, Germany) attached to an Axiovert 100 microscope; a water-immersion C-Apochromat objective was used to detect fluorescence dyes. The excitation/emission wavelengths for elastase, MPO, and Hoechst 33342 were 650/665 nm, 495/515 nm, and 528/617 nm, respectively. 18
Preparation of neutrophil extracellular traps and assay of MPO-DNA, NE-DNA, and cell-free (cf)–DNA
Freshly isolated PMNs diluted to the designated densities were incubated in microfuge tubes with or without 25 nM PMA at 37°C in 5% CO2/95% air for 2 h. Next, the PMN suspensions were washed 3 times with PBS by centrifugation at 300 × g for 5 min to remove PMA. After the third wash, the PMNs were resuspended with fresh phenol red-free RPMI-1640 medium containing 2 mM L-glutamine supplemented with 2% heat-inactivated FBS and further incubated for 4 h for the subsequent co-culture with HPAECs or for the measurement of MPO-DNA, NE-DNA, and cf-DNA levels in the medium.
Supernatant obtained from the PMN suspension was collected and then PMA was added. Subsequently, the suspension was incubated with PMA for 2 h, or with PMA for 2 h then without PMA for 4 h. Medium levels of MPO-DNA and NE-DNA were measured by enzyme-linked immunosorbent assay (ELISA), as described previously. 19 In brief, quantitative detection of MPO-DNA and NE-DNA was performed by sandwich ELISA with anti-MPO (Merck Millipore Corp., Burlington, MA, USA; catalog #07-496) and anti-NE (Merck Millipore Corp.; catalog #481001) monoclonal antibodies and a peroxidase-conjugated anti-DNA monoclonal antibody (Roche Diagnostics, Indianapolis, IN, USA; Cell Death Detection ELISA #1154467500: bottle 2). The wells of microtiter strips were coated with specific monoclonal antibodies for MPO and NE to capture MPO-DNA and NE-DNA, respectively. A peroxidase substrate (2,2'-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)) was added, which reacted with bound peroxidase to yield a soluble green product that was detected at 405 nm. Absorbance was proportional to the amount of bound horseradish peroxidase-labeled anti-DNA monoclonal antibody, and results were expressed in arbitrary units.
The medium level of cf-DNA was quantified with the Quant-iT PicoGreen dsDNA assay (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. In brief, calf thymus DNA standards (0–2 µg/mL) were diluted with Quant-iT PicoGreen reagent and incubated for 2 min at room temperature before measurement of fluorescence to create a standard curve. Data obtained from samples were then compared with the standard curve, and the results were expressed in ng/ml. Fluorescence intensity (reflecting the DNA content) was measured using a QUBITR 2.0 Fluorometer (Life Technologies), with excitation at 485 nm and emission at 538 nm.
Analysis of endothelial cell monolayers for permeability
The incubation culture plates were composed of two chambers. The base of the collagen 1-coated upper chamber was a sieve with a 0.45-μm pore size (Biocoat; Corning, Bedford, MA, USA). We used 24-well microplates for the lower chamber. To allow a confluent monolayer to grow, the culture plates were incubated at 37°C in 5% CO2/95% air for 3 days, during which time the growth medium was changed every day. PMA-stimulated PMNs were added at a ratio of 4:1 onto monolayers and were co-cultured before permeability was tested. This ratio was within the range used by others.16,20 The apparent permeability coefficient of albumin (Pa) in the HPAEC monolayer was assessed as the filtration velocity of fluorescein isothiocyanate (FITC)–labeled albumin from the upper to the lower chamber. 21 We added FITC-labeled FBS medium to the upper chamber to a final concentration of 0.4 mg/mL and growth medium containing 1% FBS to the lower chamber. After co-culture in the incubator for 4 h, samples were collected from the lower and upper chambers, and the fluorescence intensities were measured with a fluorescence microplate reader (SpectraMax; Molecular Devices, Sunnyvale, CA) using excitation and emission wavelengths of 490 and 525 nm, respectively. Albumin concentrations were determined with a standard curve, and Pa was calculated as Pa = [A]/t × 1/A × v/[L], where [A] was the albumin concentration in lower chamber; t, the time in seconds; A, the area of the membrane in cm2; V, the volume of the lower chamber; and [L], the albumin concentration in the upper chamber.
Immunofluorescence staining for VE-cadherin and F-actin
In total, 5 × 104 HPAECs were seeded on the upper chambers of 0.45-μm cell-culture inserts and cultured for 3 days to allow the growth of a confluent monolayer. PMA-stimulated PMNs were added at a ratio of 4:1 onto monolayers and were co-cultured before immunofluorescence staining. The monolayer was pretreated with 10 U/ml DNase 22 and 50 μg/mL NE inhibitor 23 10 min before incubation with PMNs. The monolayer was also pretreated with 1 mM chloro (CL)-amidine. 24 After co-culture with PMNs in the incubator for 4 h, cells were washed in cold PBS and fixed in 4% paraformaldehyde. Non-specific binding of antibody was blocked with 1% FBS in PBS for 30 min. Thereafter, cells were stained with VE-cadherin antibody conjugated with orange fluorescent phalloidin (Abcam) and with F-actin antibody conjugated with green fluorescent phalloidin (Abcam) at 4°C overnight and then washed with PBS. Finally, Hoechst 33342 was used to stain nuclei. Samples were examined with a Zeiss CLSM 710 system. VE-cadherin expression around the cell was counted in six different fields in each slide, and mean fluorescence intensity was quantified with Zen image software (version 3.1; Zeiss).
Neutrophil extracellular trap inhibition
To elucidate the contribution of NETs during co-culture with PMA-activated PMNs, we assessed the effect of inhibitors of NET synthesis. One critical step during NET synthesis is chromatin decondensation, which is associated with histone citrullination, a process catalyzed by the enzyme peptidyl arginine deiminase 4 (PAD4). The backbones of NETs are made of extracellular cf-DNA decorated with toxic compounds such as NE, MPO, and histones. Ten minutes before incubation with PMNs, the monolayer was pretreated with 10 U/ml DNase; 50 μg/mL 1-(3-methylbenzoyl)-1H-indazole-3-carbonitrile, a specific NE inhibitor; or 1 mM CL-amidine, a pan PAD inhibitor. At this concentration, no effects of DNase, CL-amidine, or NE inhibitor were seen on albumin permeability, VE-cadherin expression, or cytotoxicity of HPAECs, as assessed using a fluorescence microplate reader, image processing software, and MTT assay (data not shown).
Statistical analysis
All statistical analyses were performed with SigmaPlot software, version 14 (Systat Software Inc., CA, USA). Using mean differences and corresponding SDs of albumin permeability and VE-cadherin expression in HPAECs and parameters of NET formation including cf-DNA, MPO-DNA, and NE-DNA, we calculated that a sample size of 5–7 was needed to find differences with a two-sided confidence interval of 0.95 and a desired power of 0.8. Continuous variables are shown as the mean with SD. For group comparison, a one-way analysis of variance was used, followed by a post hoc Holm-Sidak test.
Results
Analysis of neutrophil extracellular trap formation
Medium levels of cell-free DNA, neutrophil elastase-DNA, and myeloperoxidase-DNA before and after activation of neutrophils by phorbol myristate acetate.
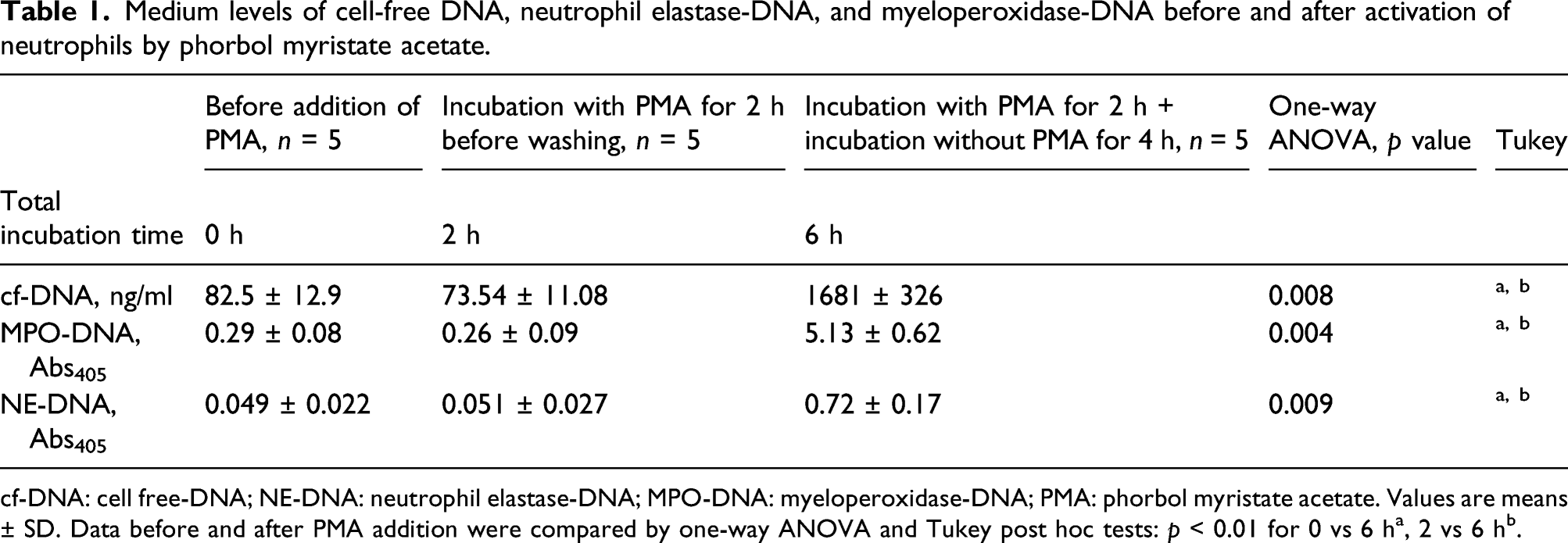
cf-DNA: cell free-DNA; NE-DNA: neutrophil elastase-DNA; MPO-DNA: myeloperoxidase-DNA; PMA: phorbol myristate acetate. Values are means ± SD. Data before and after PMA addition were compared by one-way ANOVA and Tukey post hoc tests: p < 0.01 for 0 vs 6 ha, 2 vs 6 hb.

Detection of neutrophil extracellular traps (NETs) by immunolabeling. Representative images showing direct immunofluorescence staining of DNA (blue), myeloperoxidase (MPO, green), and neutrophil elastase (NE, red) in neutrophils and NET structure before (a) and after addition of phorbol myristate acetate (PMA; b and c). Neutrophils were incubated with PMA for 2 h (b) and then washed three times to remove PMA and further incubated without PMA for 4 h (c). The top row shows higher magnification views of the selected areas in the lower row. Scale bars: 50 μm. n = 4–6.
Neutrophil extracellular traps influence permeability in human pulmonary artery endothelial cells
To evaluate the effects of activated PMNs on permeability across the HPAEC monolayer, we assessed Pa in a model of HPAECs co-cultured with PMNs activated by PMA. After the addition of PMNs activated by PMA, Pa significantly increased when compared with co-culture with PMNs without PMA stimulation (Figure 2). This increase in permeability was significantly reduced by pretreatment with CL-amidine, DNase, or NE inhibitor (Figure 2). The apparent permeability coefficients of albumin (Pa) in a human pulmonary artery endothelial cell (HPAEC) monolayer induced by co-culture with phorbol myristate acetate (PMA)-activated neutrophils. Fluorescein isothiocyanate (FITC)–labeled albumin in the lower chamber with permeation through the HPAEC monolayer co-cultured with and without PMA-treated neutrophils in the presence and absence of DNase, a neutrophil elastase (NE) inhibitor, and a CL-amidine. Values are means ± SD; n = 4. **p < 0.01 vs without PMA. †p < 0.05; ††p < 0.01 vs with PMA.
Neutrophil extracellular traps affect human pulmonary artery endothelial cell cytoskeletal and VE-cadherin
Endothelial adherens junctions play a key role in maintaining the integrity of cell-cell junction structures. We examined the adherens junction component VE-cadherin under co-culture conditions. The distribution of VE-cadherin in HPAECs co-cultured with stimulated PMNs was significantly altered compared with HPAECs co-cultured with unstimulated PMNs: the latter showed a straight, linear distribution of VE-cadherin between the cells (Figure 3(a)); however, almost all VE-cadherin expression disappeared in HPAECs co-cultured with stimulated PMNs (Figure 3(b) and (f)). When HPAECs were pretreated with DNase (Figure 3(d)), a PAD4 inhibitor (Figure 3(e)), or an NE inhibitor (Figure 3(c)), an interrupted (Figure 3(e), dashed arrows) and a zig-zag (Figure 3(c) and (d), white arrows) redistribution patterns appeared in the HPAECs and their VE-cadherin expression was higher than that of HPAECs co-cultured with stimulated PMNs (Figure 3(f)). Many cytoplasmic bodies visualized by double-immunofluorescence staining were observed in cells co-cultured with PMA-activated neutrophils (Figure 3(b)–(e)). Detection of changes in vascular endothelial (VE)–cadherin and cytoskeleton F-actin of human pulmonary artery endothelial cells co-cultured with phorbol myristate acetate (PMA)–treated neutrophils in the presence or absence of an inhibitor of neutrophil extracellular traps (NETs). (A–E): Intercellular junctions were evaluated by detecting the adherens junction protein VE-cadherin (red). The cytoskeleton was evaluated by detecting F-actin (green). DNA was stained using Hoechst 33248 (blue). Brown arrows indicate stress fibers. White arrows indicate a zig-zag pattern; dashed arrows indicate an interrupted pattern. NET inhibition was performed by DNase, a CL-amidine, and a neutrophil elastase (NE) inhibitor. Scale bar: 20 μm. (f): The immunofluorescence staining intensities of VE-cadherin, expressed as mean fluorescence intensity (MFI). MFI was quantified using Zen image software. n = 5–6. **p < 0.001 vs without PMA. ††p < 0.001 vs with PMA.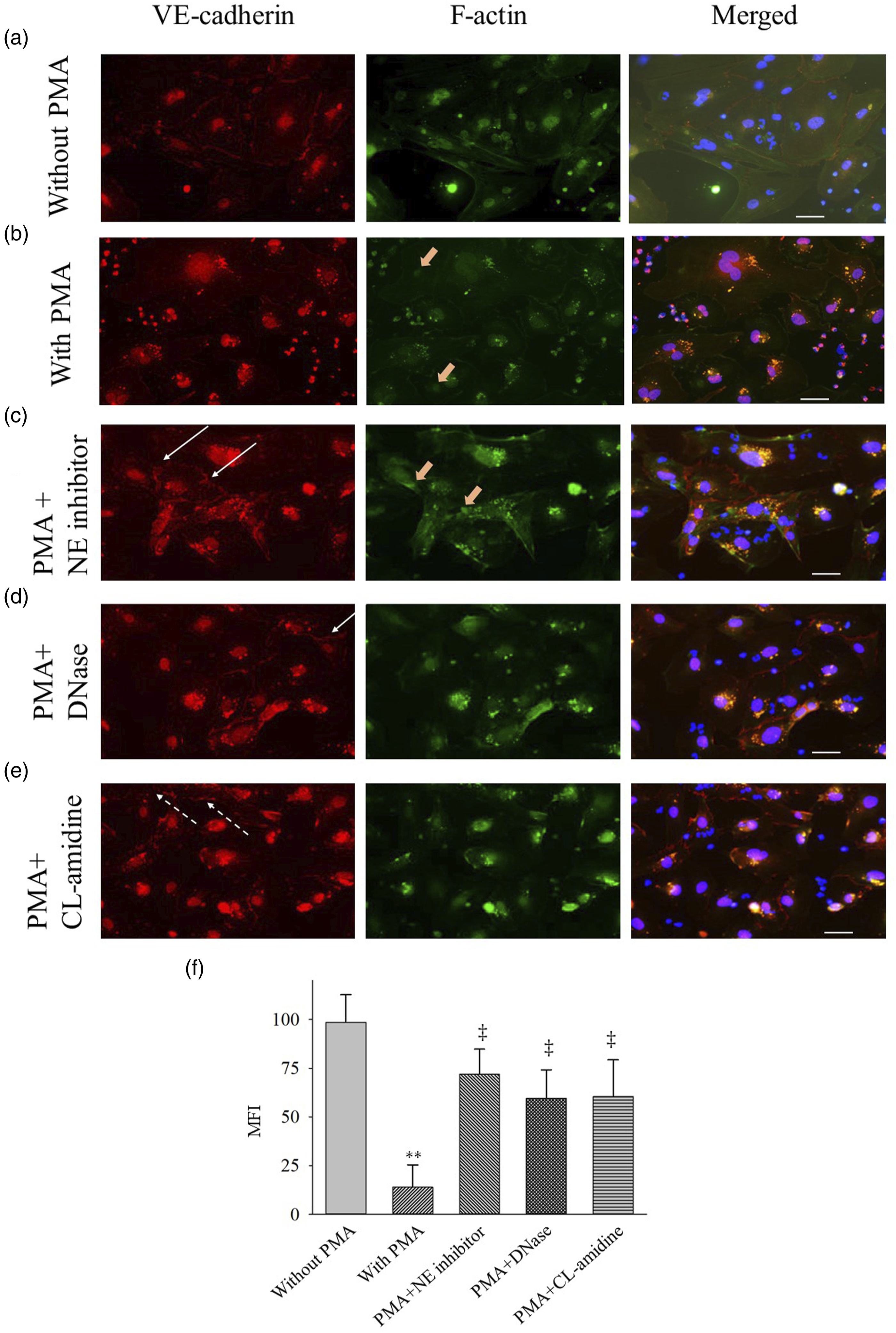
Cellular actin filament alignment was assessed by F-actin staining. HPAECs co-cultured with stimulated PMNs caused the remodeling of the endothelial actin cytoskeleton, that is, actin fibers moved from the cortical rim and were collected and condensed as intracellular stress fiber (Figure 3(b) and (c)). The alteration in F-actin in HPAECs co-cultured with stimulated PMNs was partially prevented when HPAECs were pretreated with DNase, PAD4 inhibitor, or NE inhibitor (Figure 3(c)–(e)).
Discussion
This in vitro study showed that co-culture with stimulated PMNs increases the permeability of the HPAEC monolayer and causes cytoskeleton remodeling and alterations of VE-cadherin at the cell-cell junction. These effects were prevented by pretreating HPAECs with DNase, a PAD4 inhibitor, and an NE inhibitor, suggesting that formation of both NETs and NET components affects HPAEC barrier function.
One of the key features of ARDS is the dysregulation and recruitment of activated neutrophils to the lung microvasculature, interstitium, and alveolar space.16,25 This excessive neutrophil activation and accumulation induces not only increased formation of NETs but also increased release of proinflammatory mediators,26,27 degradation of glycocalyx,28,29 and increased formation of reactive oxygen species.26,30 This study shows that both NETs and NET components are factors that probably account for the lung endothelial barrier dysfunction caused by activated neutrophils. Our results are in line with the data recently published by Lv et al., 31 although that group used a cell line of different origin. With the direct visualization xCELLigence system, they showed that lipopolysaccharide (LPS)-activated neutrophils (referred to as NETing neutrophils) caused homologous lung epithelial injuries in a time- and number-/concentration-dependent manner. Together, they showed that both pulmonary endothelial and epithelial cells are injured directly by NETs, a process that might be involved in the pathogenesis of ARDS.
DNase has been proposed as a protective agent against NET toxicity in vivo14,32–34 and in vitro.22,31 Digestion of NETs by DNase significantly ameliorated tissue injury and the degree of systemic and local inflammation triggered by LPS in a mouse model of ARDS. 32 Several studies have showed the participation of excess NET formation in pathologic processes in SARS-Cov-2–induced ARDS.11,35 Therefore, a study is currently being performed in patients with SARS-Cov-2–induced ARDS to evaluate the therapeutic effects of administering intratracheal DNase to digest NETs structures. 36 NE is also a component of NETs and plays a central role in their toxicity. 37 In this study, we showed that treatment with an NE inhibitor improved the degree of permeability compared with the condition without an NE inhibitor.
Our findings on the protective effect of DNase and an NE inhibitor on NET-related endothelial barrier dysfunction are consistent with a previous report, which showed that acid aspiration–induced ARDS in mice was markedly attenuated by administration of DNase and an NE inhibitor. 33 The study found that NETs are involved in the pathogenesis of acid aspiration–induced ARDS in both humans and animal models. 33
The contribution of NETs to the increased permeability of the HPAEC monolayer was also confirmed by the peptidylarginine deiminase (PAD) inhibitor CL-amidine, which dismantles or inactivates existing NETs by a different mechanism.38,39 PAD4 is important for chromatin decondensation during NETosis because it modifies histone charges through citrullination.40,41 A PAD inhibitor was successfully used in several animal studies to prevent NET-associated pathogenesis. PAD4−/− deficiency in mice protected against cecal ligation and puncture-induced septic shock42,43 and bacterial pneumonia–induced lung injury. 14 Experimental animal ARDS in response to combined hemorrhage and sepsis was less severe in PAD4−/− mice than in wild-type mice, providing evidence to support the notion that PAD4/NETs contribute to lung injury. 43
To examine the precise pathogenetic role of NETs in the increased permeability of the HPAEC monolayer, we studied the integrity of HPAEC barrier function, which is essential for maintaining oxygenation of the lung. The integrity of the adherens junction is particularly important for regulating paracellular permeability via homophilic adhesions between VE-cadherin molecules,44–46 actin cytoskeleton remodeling,46–48 and intercellular signaling. 46 The changes in endothelial VE-cadherin and actin structures observed in HPAECs co-cultured with PMA-activated PMNs led us to hypothesize that MPO, NE, and cf-DNA released by NETs damage the endothelial cells by binding to factor XII and toll-like receptors 2 and 4,22,36,49,50 which may result in the loss of adherens junctions. Actin cytoskeleton remodeling, including stress fiber formation, causes the cells to contract, increasing permeability.46–48 Meegan et al. showed that citrullinated histone 3 induced endothelial barrier dysfunction characterized by reorganization of the cytoskeleton with increased F-actin stress fibers. 51 Jerke et al. showed that neutrophil serine proteases including neutrophil elastase and cathepsin G disturbed the endothelial cell cytoskeletal architecture and barrier function. 52 Further experiments are required to clarify which components of NET-related proteins contribute to endothelial cell damage.
The involvement of NETs in endothelial barrier dysfunction was confirmed by the reverse effects of inhibiting NET formation with PAD4 inhibitor or NE inhibitor and of increasing the degradation of the structure of NETs with DNase. The alterations of the adherens junction and cytoskeletal rearrangement were studied in several animal models and in patients with ARDS. In an experimental mouse ARDS model, Saponznikov et al. 53 examined the early disruption of the alveolar-capillary barrier. They showed that the adherens, tight, and gap junction protein families were rapidly diminished in the ARDS model. Similarly, pulmonary VE-cadherin protein levels were reduced in the LPS-induced lung injury model54,55 and in lung specimens collected from patients with ARDS and sepsis. 56
Previous studies demonstrated that plasma levels of circulating cell-free NETs were higher in patients with bacterial14,15 and viral11,16 pneumonia-associated ARDS, blood transfusion–associated ARDS, 57 and remote lung injuries 58 than in individuals without ARDS. In addition, the levels of circulating cell-free NETs correlated to the severity of ARDS, indicating that NETs were accelerated in ARDS and may play a central role in the pathogenesis of ARDS. NET-targeted therapies, such as those inhibiting de novo NET synthesis or accelerating the degradation of preformed NETs, are potential therapeutic avenues to be explored in further investigations.
This study has some limitations. First, we used only PMA to induce NETs. Even though PMA is consistently reported to be a NET inducer, it is not physiologically significant because it does not activate physiological processes in vivo. Therefore, the effects of other (physiological) NET inducers, including live bacteria and fungi, should be studied. Second, many cytoplasmic bodies were observed in cells co-cultured with activated neutrophils. Because cytoplasmic bodies were only observed in endothelial cells co-cultured with stimulated neutrophils, leukocyte/endothelial interaction including amplified reactive oxygen species production and neutrophil migration through endothelial junction may be related to the formation of cytoplasmic bodies. Third, an in vitro setup cannot completely reflect the in vivo situation. Other immune cells, platelets, complements, and mediators are expected to be involved in an immune and inflammatory response in vivo. Further studies are needed to confirm the extent to which NETs contribute to lung endothelial cell injuries.
Conclusions
This in vitro study indicates that lung endothelial barrier functions are altered by NET formation and the extracellular release of NET components. Hyperpermeability, cytoskeleton remodeling, and alteration of VE-cadherin in cell-cell junctions are prevented by inhibition of NET formation and increased degradation of NET structure. NETs would be involved in the increased vascular permeability of the lung, which seems to be a common feature in ARDS of various etiologies. Thus, this study provides insights that may help generate novel approaches for medical interventions.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was funded by a JSPS KAKENHI Grant-in-Aid for Scientific Research C (17K11598).
Author contributions
Study conception and design: NT. Data collection: HM, NT, MAH, and MMI. Analysis and interpretation of data: HM, NT, MAH, and MMI. Writing the manuscript: HM and NT. Critical revision: HM and NT. All the authors read and approved the final manuscript.
Originality
The authors declare that no significant parts of the data reported in this manuscript have been published elsewhere.
Ethics approval and consent to participate
Written informed consent was obtained from the healthy volunteers who participated in the study. The study protocol conformed with the ethical guidelines of the 1975 Declaration of Helsinki and was approved a priori by the Institutional Review Board of Aichi Medical University.
Availability of data and material
The data that support the findings of this study are available from the corresponding author, NT, upon reasonable request.