
Abstract
The system of urotensin II (UII) and its receptor (UT) (or: UII/UT system) mediates hepatic immune inflamed injury in acute liver failure (ALF) with autophagy inhibition. However, it is unknown whether the system has an effect on liver autophagy in ALF. In this study, we attempted to explore hepatic autophagy response in ALF through blocking the UII/UT signal. Autophagy-related genes were examined in the liver tissues of lipopolysaccharide (LPS)/d-galactosamine (D-GalN)-induced ALF after pretreatment of UT receptor specific antagonist urantide. And then, the levels of autophagy- and apoptosis-related genes were assayed in LPS-stimulated KCs via urantide pretreatment. We found that the expressions of hepatic autophagy related genes, including Beclin-1, Atg5, Atg7, LC3 and p62 mRNA, and LC3 II and p62 protein, were significantly downregulated in LPS/D-GalN-induced ALF mice; but they were not affected by pretreatment of urantide, a special UT receptor antagonist. To probe inflammatory mechanisms of the UII/UT system, we further investigated the effect of the system on Kupffer cells (KCs), the innate immune cells in liver. We found that urantide pretreatment significantly inhibited production of inflammatory injury molecules including TRAF6 and ROS in LPS-stimulated KCs. LPS stimulation induced LC3 and p62 mRNA and LC3 II and p62 protein expression in KCs. After urantide pretreatment, LC3 and p62 mRNA and LC3 II protein were downregulated, while p62 protein was upregulated in LPS-stimulated KCs. In addition, antiapoptotic protein Bcl-2 inhibition and proapoptotic protein cleaved caspase-3 increase were observed in LPS-stimulated KCs, and the effects were enhanced after urantide pretreatment in the study. We conclude that liver injury mediated by the UII/UT system is possibly associated with the activation of autophagy-related and apoptosis-resisted pathways of KCs in ALF.
Introduction
Acute liver failure (ALF) is a severe liver disease associated with immunological inflammatory response. Urotensin II (UII), a cyclic eleven-amino acid polypeptide, and its specific G-protein coupled receptor GPR14 (UT) are collectively called the UII/UT system. 1 UII/UT system has a variety of physiological and pathological activities. 2 In recent years, it has been discovered that the UII/UT system plays an important role in inflammatory diseases. 3 Enhanced expression of UII/UT was demonstrated in the liver of patients with ALF. 4 Our previous study observed that UII levels increased in the very early stage of LPS/D-GalN-induced ALF, which mediated the proinflammatory cytokines secretion such as TNF-α and IL-1β. 5 Furthermore, an administration of urantide (UT specific antagonist) can not only block UII/UT signal transduction, but also protect the mice from hepatocyte apoptosis, liver inflammation and animal death caused by LPS/D-GalN challenge. 6
The UII/UT system is mainly expressed in Kupffer cells (KCs) in liver tissues with ALF. 6 KCs are the resident macrophages in the liver, which is an important component of the innate immunity system and contributes to the resistance of exogenous toxins and infectious microorganisms from the portal vein. 7 In the liver, KCs are the major source of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β, which facilitate the inflammatory hepatic injuries and finally result in ALF.8,9 Our previous study has demonstrated that the UII/UT system mediates the over-expressions of proinflammatory cytokines through p38-MAPK and NF-κB pathways in LPS-stimulated KCs. 10 In general, the UII/UT system may be the trigger of LPS/D-GalN-induced liver inflammatory injuries. 5
Liver injury is another coin face of hepatic protective inability in ALF. However, the effect of the UII/UT system on liver protection in ALF remains unclear. As a liver protective mechanism, autophagy is the process of cellular self-eating, and has long been recognized as a key protein degradation pathway. Cytoplasmic components including proteins and organelles are sequestered by autophagosome, and are then transferred to the lysosome for degradation, particularly during starvation or stress. 11 Evidence has indicated that autophagic degradation promotes the recycling of cellular nutrients and elimination of toxicity, thereby enabling cell survival. 12 More than 30 Autophagy-related proteins (ATGs) are involved in this four-stage progress: initiation, nucleation, expansion and degradation. 11 Among these regulatory genes, Beclin-1 participates in the nucleation process of autophagy, while Atg5 and Atg7 are involved in the expansion of autophagy. Some autophagy related proteins catalyze microtubule associated protein light chain I (LC3 I) to bind covalently to phosphatidylethanolamine to form autophagic endometrial specific LC3 II. LC3 II can promote the extension of autophagic membrane. P62 protein (also known as SQSTM1 protein) is an important autophagic receptor protein, which main function is helping LC3 II to be in combination with the degrading target protein or damaged organelles. Insufficient function of autophagic lysosomes leads to p62 accumulation. Hence, the intracellular content of p62 can be used to assess the activity of autophagic lysosomes.11,13 Many liver diseases, such as alcoholic liver disease and nonalcoholic fatty liver, are demonstrated to be associated with autophagy. 14 In addition, recent studies have discovered that the level of autophagy is obviously abnormal in the pathogenesis of ALF. 15 Inhibition of autophagy can aggravate the liver damage, whereas the enhance of autophagy reduces the damage and the animal mortality in ALF.15,16 This suggests that autophagy demonstrates a protective property in ALF.
Therefore, this study explores the effect of the UII/UT system on the expressions of autophagy-related genes in the liver tissues of ALF. In addition, in order to further understand the role of autophagy in the UII/UT system-mediated hepatic inflammatory damage, it examines the expressions of autophagy- and apoptosis-related genes under LPS-stimulated KCs. Such a study may illuminate the underlying mechanisms where liver injury is mediated by the UII/UT system.
Materials and methods
Materials
Methods
Animal experimental
The method was performed according to our previous article. 6 Mice were intravenously injected before ALF induction with 0.6 mg/kg urantide dissolved in 100 μl NS (urantide group, n = 6) or with 100 μl of normal saline (NS group, n = 6). After 30 min of the injection, mice (NS and urantide groups) were challenged by an injection intraperitoneally with 200 μl of NS or with 800 mg/kg GalN and 50 mg/kg LPS dissolved in 200 μl of NS (LPS/GalN group, n = 6 and urantide + LPS/GalN group, n = 6). Liver tissues and blood were collected 6 h after LPS/GalN challenge.
Isolation of primary KCs
Rat KCs were isolated following our previous article. 17 Simply, rats were intraperitoneally anesthetized with sodium pentobarbital (40 mg/kg) and 1 ml of heparin. After opening the abdomen and the ligation of the suprahepatic inferior vena cava, D-Hanks’ solution was slowly injected through the portal vein, and the hepatic inferior vena cava was cut open to allow blood and perfusion fluid to drain freely. When the color of the liver turned white, the solution was switched to IV collagenase digestion solution. The liver tissues were dissected and torn apart with IV collagenase solution, and then passed through a 200-mesh filter. The collected cells were subjected to discontinuous Percoll density gradient centrifugation. The membranous cell layer was aspirated and washed with HBSS, and then added to RPMI 1640 medium. KCs were purified via washing away non-adherent cells after 1–2 h of incubation. The purified KCs were identified using ink and ED2 staining.
KC experimental
KC treatment was performed according to our previous article. 10 Simply, KCs were incubated in 6-well plates with 4 × 106 cells/well. After 24 h, cells were washed with PBS (phosphate buffered solution), and serum-free medium was added, followed by treatment using urantide. The concentration of urantide was 20 nM, and the dosage and usage were applied as previously described. After 30 min, LPS (20 μg/ml) was used to stimulate the cells. 6 h after LPS stimulation, cells, and supernatants were collected for experiment. The experiment had four groups including control, urantide, LPS, and urantide + LPS (six animals each group).
Real-time PCR assay
Method of Real-time PCR was described in our previous article. 17 Briefly, liver tissues and KCs were lysed with Trizol to extract total RNA, which was used as the template for the synthesis of first-strand cDNA. The resulting cDNA was then used as the template, Taq DNA polymerase and the upstream and downstream primers of the target genes were added, and PCR was amplified according to the manual. The primers were designed and synthesized by Shanghai Sangon Biotech (China); the primer sequences and product sizes used in gene assay are shown in Table 1. Real-time PCR was carried out according to the kit instructions. The reaction system was added with 2× Premix Ex TaqTM II, sense and antisense strand primers, ROX Reference Dye II, and DNA templates, and was amplified in the ABI 7500 instrument by the following two-step method: Step One: 95°C for 30 s, 1 cycle; Step Two: 95°C for 5 s and 60°C for 34 s, 40 cycles. GAPDH was used as the internal reference.
Primer sequences used for PCR.

Western-blot analysis
Method of western-blot analysis was described in our previous article. 17 Briefly, protein was extracted from liver tissues and KC cells in RIPA buffer together with phosphatase and protease inhibitors. 50 μg of protein was collected, boiled in Laemmli buffer for 10 min, and separated on 12% polyacrylamide gel; then it was transferred to a PVDF membrane. Target proteins were detected by Western blot with the specific first antibody (including β-actin, TRAF6, LC3, p62, Bcl-2, and cleaved caspase-3) in blocking buffer overnight at 4°C. After washing, the membranes were further probed with the horseradish peroxidase-conjugated secondary antibody (1:1000) in blocking buffer for 1 h at room temperature. The western chemiluminescent HRP substrate (Millipore Corporation, Billerica, MA, USA) was used for chemiluminescence development. Finally, the quantitative results of western blots were assessed by ImageJ to make comparisons between different groups.
Measurement of ROS production
ROS measurement was performed according to kit instruction and referred to the article. 18 ROS levels were determined by measuring the oxidative conversion of 2′, 7′-dichlorofluorescin diacetate (DCFH-DA) to the fluorescent compound dichlorofluorescin (DCF). In brief, the KCs that were seeded in 6-well plates and underwent various treatments (details in KC experimental) for the indicated time intervals were incubated with DCFH-DA solution (15 µM, final concentration) for 0.5 h at 37°C. After washing, Inverted fluorescence microscope (Leica DMIRB IX51) was used to observe the fluorescent signals. Then, KC cells were trypsinized and collected via centrifugation at 500g for 5 min. The collected cells were suspended in 500 ul RPMI 1640 media. Cell concentration was detected by Automatic cell counter (Bio-Rad TC10). DCF fluorescence was determined at 485 nm excitation and 520 nm emission using a Luciferase detector (Promega Glomax 20/20). The mean fluorescence intensity was used to reflect the ROS levels.
Statistical analysis
The results are expressed as the means ± standard deviation of six independent experiments and intergroup comparison is assayed by one-way analysis of variance (ANOVA). The SPSS22.0 statistical software has been used for statistical analysis, and p < 0.05 indicates a statistically significant difference.
Results
Hepatic injury was mediated by the UII/UT system in LPS/D-GalN-challenged Mice
Identical to the results of our previous study, 6 this experiment has witnessed a precipitous increase in the serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) after LPS/D-GalN injection (all p < 0.01 vs control mice); however, the levels have decreased significantly via pretreatment of urantide, the special receptor anagonist of UT in the LPS/D-GalN-challenged mice (all p < 0.01) (Figure 1). It suggests that LPS/D-GalN-induced liver injury can be mediated by the UII/UT system.
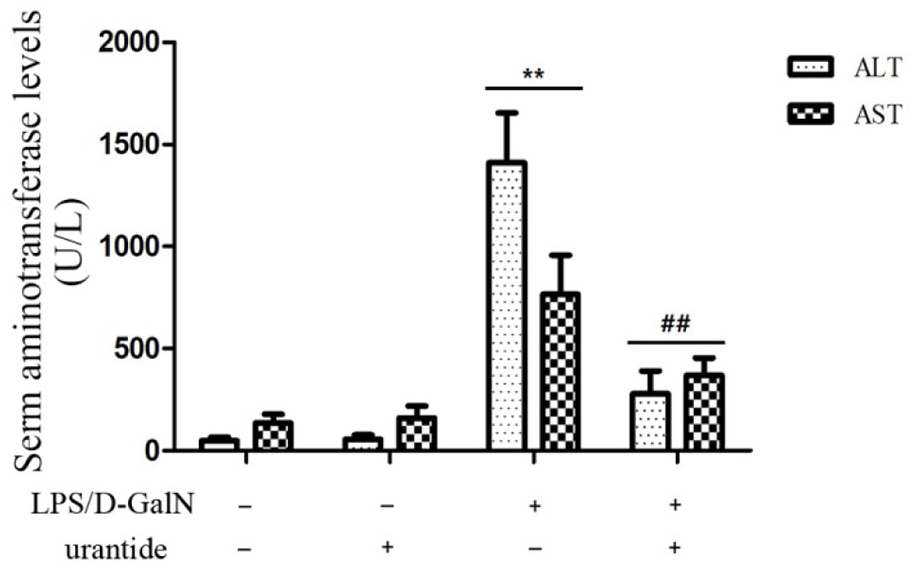
Serum levels of ALT and AST in mice Mice were treated with urantide or vehicle 0.5 h before LPS/D-GalN injection. (n = 6 each group). *p < 0.05 and **p < 0.01 versus control mice (urantide(−)LPS/D-GalN(−)); #p < 0.05 and ##p < 0.01 versus LPS/D-GalN-challenged mice (urantide(−)LPS/D-GalN(+)).
Inhibition of hepatic autophagy-related pathway was unaffected by urantide in LPS/D-GalN-challenged mice
To investigate the relationship between UII/UT system and liver autophagy in ALF, we measured hepatic autophagic related genes in LPS/D-GalN-challenged mice using UT antagonist urantide. Our results indicate that the hepatic mRNA levels of Beclin-1, Atg5, Atg7, LC3, and p62, and the protein levels of LC3 II and p62 decreased in LPS/D-GalN-challenged mice (all p < 0.01). However, compared with the ALF mice, urantide-pretreated mice presented no statistical difference in mRNA and protein levels of these autophagic genes in the liver tissues (all p > 0.05) (Figure 2). It suggests that LPS/D-GalN-induced autophagic pathway inhibition may not be affected by the UII/UT system in mouse liver.
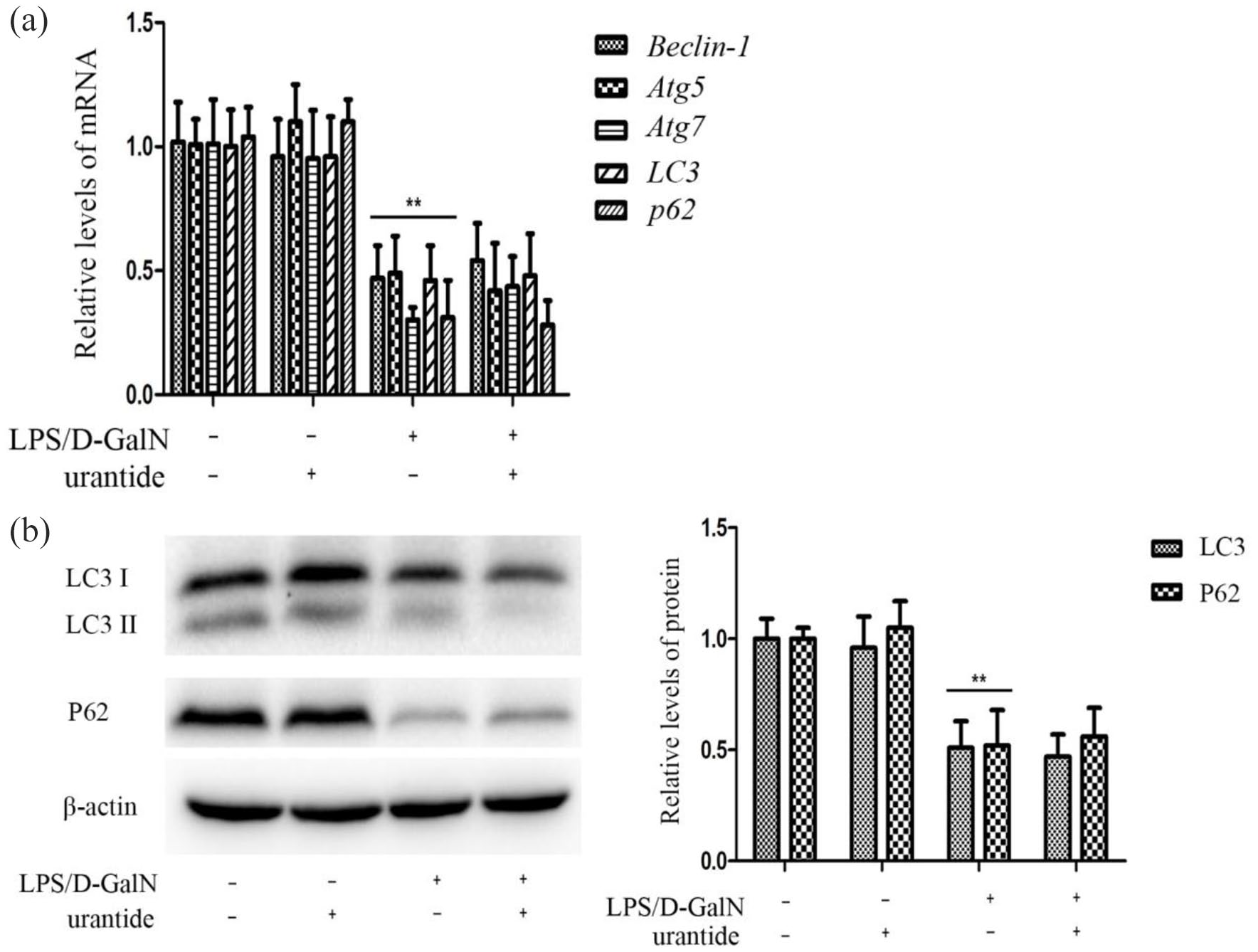
Expression of Beclin-1, Atg5, Atg7, LC3, and p62 mRNA and LC3 and P62 protein in mouse liver tissues. Mice were treated with urantide or vehicle 0.5 h before LPS/D-GalN injection. (a) Beclin-1, Atg5, Atg7, LC3, and p62 mRNA expression in liver using qRT-PCR assay. The panel shows relative expression levels of Beclin-1, Atg5, Atg7, LC3, and p62 mRNA in liver after normalization to GAPDH. Data represent means ± SD (n = 6 each group). (b) LC3 II and P62 protein expression in liver using Western blot. Left panel shows a representative picture of Western blot, and right shows relative levels of LC3 and P62 protein after normalization to β-actin. In SDS-PAGE electrophoresis, LC3 is separated to two bands, namely LC3 I and LC3 II. Because LC3 II is modified by phosphatidylethanolamine, leading to the charge change and making the migration rate faster. In this figure, the molecular weight of LC3 I is 16 kDa, and the molecular weight of LC3 II is 14 kDa. Data represent means ± SD (n = 6 each group). *p < 0.05 and **p < 0.01 versus control mice (urantide(−)LPS/D-GalN(−)); #p < 0.05 and ##p < 0.01 versus LPS/D-GalN-challenged mice (urantide(−)LPS/D-GalN(+)).
TRAF6 expression and ROS release were inhibited by urantide in LPS-stimulated KCs
As a kind of innate immune cells in liver, KCs have a pivotal role in the hepatic inflammatory and oxgen injury in the pathogenesis of ALF.19–21 In this study, we further assayed the effect of UII/UT system on TNF receptor-associated factor 6 (TRAF6), the upstream inflammatory signal of TNF-α and IL-1β, and ROS, the oxgen stress injury molecule in primary KCs (cell purity > 90% and viability > 95%, similarly hereinafter). Our results show that after LPS stimulation, the levels of TRAF6 mRNA and protein expression and ROS production of KCs were considerably increased (all p < 0.01 vs control cells), whereas urantide pretreatment significantly inhibited LPS-induced TRAF6 and ROS upregulation (all p < 0.01 vs LPS-stimulated cells) (Figure 3). This result indicates that UII/UT system mediates LPS-induced TRAF6 inflammatory signal and ROS oxygen stress in KCs.

TRAF6 expression and ROS release in KCs. KCs were treated with urantide 0.5 h before LPS stimulation: (a) TRAF6 mRNA expression was measured by qRT-PCR. The panel shows relative expression levels of TRAF6 mRNA in KCs after normalization to GAPDH. Data represent means ± SD (n = 6 each group), (b) TRAF6 protein expression in KCs using Western blot. Left panel shows a representative picture of Western blot, and right shows relative levels of TRAF6 protein after normalization to β-actin. Data represent means ± SD (n = 6 each group), and (c) ROS production by measuring the oxidative conversion of 2′,7′-dichlorofluorescin diacetate (DCFH-DA) to the fluorescent compound dichlorofluorescin (DCF). Left panel shows ROS releases visualized by Fluorescence Inversion Microscope System (200×), and right shows the relative levels of ROS production (n = 6 each group). *p < 0.05 and **p < 0.01 versus control cells (urantide(−)LPS(−)); #p < 0.05 and ##p < 0.01 versus LPS-stimulated KCs (urantide(−)LPS(+)).
LPS-induced autophagic pathway activation was inhibited by urantide in LPS-stimulated KCs
To investigate the impact of UII/UT system on the KC autophagy, we measured the expression levels of LC3 and p62 mRNA and p62 protein using urantide pretreatment in LPS-stimulated KCs. As is shown in Figure 4, the levels of LC3 and p62 mRNA, and LC3 II and p62 protein in LPS-stimulated KCs increased significantly (all p < 0.01 vs control cells), and LC3 and p62 mRNA and LC3 II protein levels decreased just as much. However, it is worth noting that, after urantide pretreatment, p62 protein levels upregulated extensively (all p < 0.01 vs LPS-stimulated cells). This result suggests that urantide can suppress LC3 and p62 gene transcription and LC3 II conversion of LC3 protein, and enhance p62 accumulation, indicating that UII/UT system mediates autophagic pathway activation in LPS-stimulated KCs.

Expression of autophagy genes in KCs: (a) LC3 mRNA in KCs by qRT-PCR. The panel shows relative expression levels of LC3 mRNA in KCs after normalization to GAPDH (n = 6 each group), (b) LC3 II and p62 protein expression in KCs using Western blot. Left panel shows a representative picture of Western blot, and right shows relative levels of LC3 II and p62 protein after normalization to β-actin. In SDS-PAGE electrophoresis, LC3 is separated to two bands, namely LC3 I and LC3 II. Because LC3 II is modified by phosphatidylethanolamine, leading to the charge change and making the migration rate faster. In this figure, the molecular weight of LC3 I is 16 kDa, and the molecular weight of LC3 II is 14 kDa. Data represent means ± SD (n = 6 each group). *p < 0.05 and **p < 0.01 versus control KCs (urantide(−)LPS(−)); #p < 0.05 and ##p < 0.01 versus LPS-stimulated KCs (urantide(−)LPS(+)).
LPS-induced apoptosis-related signal was enhanced by urantide in KCs
Autophagy can help maintain cellular homeostasis and fight against cell damage, and autophagy inhibition can aggravate cell apoptosis. 22 To investigate the effect of UII/UT system on KC apoptosis, we detected the expression levels of anti-apoptosis protein Bcl-2 and apoptosis protein cleaved caspase-3 using UT antagonist urantide in LPS-stimulated KCs. We found that LPS downregulated Bcl-2 protein levels and is much reduced by urantide (Figure 5(a)). Meanwhile, LPS upregulated cleaved-caspase3 levels and escalated after urantide pretreatment (Figure 5(b)). Therefore, according to this experiment, where urantide enhanced LPS-induced apoptosis-related pathway activation, we may reach the conclusion that the UII/UT system may mediate apoptotic inhibition in LPS-stimulated KCs.
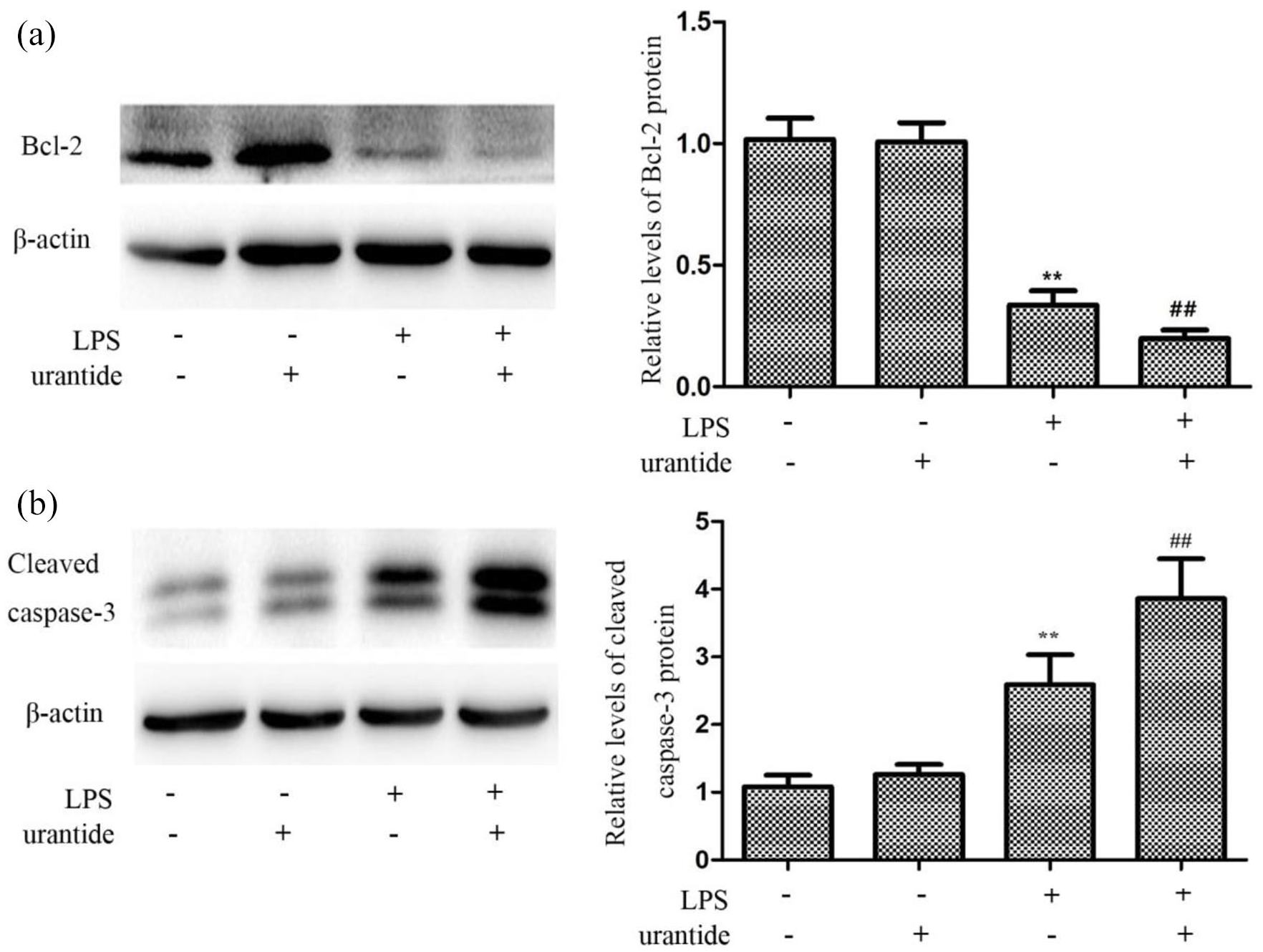
Urantide pretreatment enhanced the apoptosis in LPS-stimulated KCs: (a) Bcl-2 protein expression in KCs using Western blot. Left panel shows a representative picture of Western blot, and right shows relative levels of Bcl-2 protein after normalization to β-actin. Data represent means ± SD, (b) cleaved caspase-3 protein expression in KCs using Western blot. Left panel shows a representative picture of Western blot, and right shows relative levels of Cleaved caspase-3 protein after normalization to β-actin. In SDS-PAGE electrophoresis, Cleaved caspase-3 protein has two subunit of 17 and 12 kDa. Data represent means ± SD were showed in a representative picture of western blot and its relative levels after normalization to β-actin (n = 6 each group). *p < 0.05 and **p < 0.01 versus control KCs (urantide(−)LPS(−)); #p < 0.05 and ##p < 0.01 versus LPS-stimulated KCs (urantide(−)LPS(+)).
Discussion
The discovery of inflammatory injury effect of the UII/UT system is an important progress in immunological research in recent years. Our research team has demonstrated that the UII/UT system mediates the innate immune inflammatory response in the liver tissues in ALF mice. 10 Autophagy, a way of maintaining intracellular homeostasis, is proven to have an early transient enhancement; then it is rapidly transferred to a lasting inhibition in the liver during ALF. 15 It is also confirmed that autophagy enhancement can reduce hepatic inflammation, whereas autophagy inhibition aggravate liver injury in ALF.15,16 This suggests that autophagy plays a key role in the pathogenesis of ALF.
In this study, we further explored the ALF hepatic autophagy by using the LPS/D-GalN-challenged rat model associated with hepatic immune inflammation. In such a model, hepatic inflammation injury was very obvious at 6 h after LPS/D-GalN attack. 23 We found again that, in this experimental, serum transaminases including ALT and AST rose significantly in the experimental rats at 6 h following LPS/D-GalN injection. We also detected the levels of the autophagy related genes including beclin-1, Atg5, Atg7, p62, and LC3 in the liver tissues during this period. We found that the expression levels of the above genes were obviously downregulated in the ALF liver. This shows that many stages of hepatic autophagy are inhibited, which suggests the downregulation or loss of hepatic cell protection mechanism.
The UII/UT system is proven to have an inhibitory effect on autophagy in previous reports.24,25 It is confirmed that the UII/UT system can shear off beclin-1 and block the formation of autophagic body through activating the downstream calpain signal. 24 Chen et al. 25 reversed the autophagic decline of renal epithelial cells in diabetic mice by applying SB-657510, a UT receptor antagonist. In order to study the effects of UII/UT system on ALF hepatic autophagy, we applied a new special UT antagonist urantide to block the UII/UT signaling in the liver according our previous article. 6 However, a statistical difference was not found between urantide pretreatment and non-pretreatment about autophagy related gene levels in LPS/ D-GalN-challenged rats in this study. This indicates that the UII/UT signal system has no effect on hepatic autophagy-related pathway in ALF. To probe the inflammatory injury mechanisms of the UII/UT system, we further investigated the effect of the system on primary separated KCs, the innate immune inflammatory cells.
In the experiments, we have found that urantide application significantly inhibits TRAF6 overexpression in LPS-stimulated KCs. TRAF6 is an important signal protein of MyD88 dependent pathway in TLR4 downstream signals. LPS initiates MyD88 dependent signal and activates TRAF6 protein by binding TLR4 on cell surface, then promotes NF-κB nuclear transfer and finally induces cascading releases of proinflammatory cytokines. 26 This indicates that the inflammatory activation and release mediated by the UII/UT system is closely related to the upregulation of the inflammatory signal molecular TRAF6 in LPS-stimulated KCs. In addition, in our study, we have also discovered that, in KCs, LPS can significantly stimulate ROS production, which is suppressed by urantide pretreatment. This suggests that the UII/UT system also mediates LPS-induced ROS in KCs. Previous report has presented that TRAF6 activation can induce mitochondria to produce ROS in LPS-stimulated macrophages. 27 A unified molecular basis may exist between inflammatory effect and oxidative injury in the innate immune inflammatory cells. Therefore, the UII/UT system plays a key role in the development of hepatic immune inflammation and oxidative damage in ALF through the important innate immune cells—Kupffer cells.
The production and persistence of hepatic inflammatory and oxidative damage need the maintenance of the survival and functional activity of Kupffer cells. Recent studies have shown that cellular survival and functional activity are closely related to autophagy.12,28 However, the effect of the UII/UT system on KC autophagy has not been reported. In this study, we found that after LPS stimulation, the expressions of LC3 mRNA, p62 mRNA, LC3 II, and p62 protein increased significantly in KCs; in other words, LPS induced KC autophagy-related pathway. After pretreatment with urantide, the expressions of LC3 mRNA, p62 mRNA, and LC3 II protein were downregulated, while the expression of p62 protein was upregulated (protein accumulation due to consumption insufficiency); viz., the activity of autophagy-associated pathway was downregulated via urantide pretreatment in LPS-stimulated KCs. Thus, the conclusion that LPS-induced KC autophagy signal depends on the activity of the UII/UT system is reached from a different approach.
In this experiment, we also studied the effect of UII/UT system on KC apoptosis-related molecules by LPS stimulation. Antiapoptotic protein Bcl-2 inhibition and proapoptotic protein cleaved caspase-3 increase were observed in LPS-stimulated KCs, and the effects enhanced after urantide pretreatment in the study. The results show that UII/UT system can prevent LPS-stimulated KCs from cell apoptosis.
The afore-mentioned results are based on our preliminary experiments; therefore, there may be some limitations or deficiencies that require further studies. For example: (1) Is there a causal relationship between autophagy/apoptosis of Kupffer cells and liver immune inflammation injury? What is the mechanism? (2) How do autophagy and apoptosis of Kupffer cells affect each other? What is the role of the UII/UT system? (3) How does the UII/UT signal system affect the oxidative/inflammatory damage effect of liver tissues through autophagy/apoptosis of Kupffer cells? And etc.
Conclusion
In conclusion, our study has confirmed the inflammatory and oxidative damage mechanisms of the UII/UT system in the liver. In this mechanism, KC activation of autophagy-associated and apoptosis-resisted pathways is the key link of the UII/UT system mediated oxidative inflammation injury.
Footnotes
Author contributions
Liu LM designed, planned and wrote the manuscript. Zhong H executed the experiments, analyzed the data and wrote the manuscript. He Y, Yang X, Si QQ, Xie P and Gao DY executed the experiments.
Animal welfare
Animal treatment was in compliance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the National Natural Science Foundation of China (No.81770612, No.81070357 and No.30660066) and Science and Technology Research Projects of Shanghai Songjiang district (No.16SJGG21).
Ethics approval
Ethical approval for this study was obtained from “Shanghai JiaoTong University School of Medicine DLAS (SJUM2020002).”