
Abstract
Introduction
Acute-on-chronic liver failure (ACLF) is a common liver problem characterized by rapid hepatic deterioration in patients with chronic liver disease associated with single or multiple organ system dysfunction and a high risk of short-term mortality.1,2 Epidemiological observations in pre-existing chronic liver disease populations indicate that several triggering factors, such as hepatotropic viruses, alcoholism, bacterial infections, autoimmune hepatitis, drug-induced liver injury, gastrointestinal bleeding and Wilson’s disease flares, and other factors, are associated with an increased risk of developing ACLF in pre-existing chronic liver disease population.1,3 Among these, hepatitis B virus (HBV) reactivation is a more common insult in Asia; however, alcoholism and bacterial infections are the most frequently identified factors in America and Europe.3–5 Although improvements in the prevention, diagnosis, and treatment of ACLF have improved the clinical features and survival outcomes of patients with the disease, the 28-days mortality rate remains ≥20%, worldwide.6,7
The definitions of ACLF vary across different regions, including Europe, America, and Asia Pacific, owing to differences in population characteristics. However, despite this, systemic inflammation plays a vital role in the current pathomechanistic paradigm of organ failure development and is a characteristic feature of patients with ACLF.4,5 Systemic inflammation induces the secretion of various proinflammatory cytokines and chemokines, recruits more immune cells to the liver, and causes extrahepatic organ failure by stimulating NO production. This exacerbates pre-existing circulatory dysfunction and activates the immune system.5,7,8 Notably, during ACLF development, systemic inflammation also damages the immune system, causing to immune-mediated damage to the liver tissue, massive hepatocyte necrosis, and liver failure.3–5 For example, patients with ACLF have more intense systemic inflammation, as indicated by high levels of inflammatory response indicators such as leukocyte count, C-reactive protein, and plasma cytokines, which closely correlate with the syndrome severity.4,9–12 In addition, the levels of inflammatory factors linked to the cytokine release syndrome in the blood, including interleukin (IL)-6, IL-8, IL-10, and tumor necrosis factor-α, are significantly increased in patients with ACLF than in those without ACLF.4,13 These inflammatory factors are the hallmarks of ACLF, making it a distinct clinical syndrome. Moreover, they contribute to organ failure and influence the disease course, causing an increased risk of short-term mortality in patients with ACLF.12–15 In an untargeted metabolomic study, the severity of systemic inflammation correlated directly with blood metabolite accumulation, indicating significant alterations in metabolic dysfunction, which may be associated with the organ failure. 14 The higher the baseline levels of inflammatory factors in the blood, the larger the number of organ failures at enrollment. 14 Inhibition of mitochondrial energy production may have particularly contributed to organ failure. 14 In most ACLF cases, activation of the innate immune system by pathogen-associated and damage-associated molecular patterns, which trigger various signaling pathways that are activated by several inflammatory factors, is believed to play a vital role in the inflammatory response, which is related to the molecular pathogenesis of ACLF.12,14–17 For example, in ACLF rats, a significant immune dysregulation was observed at the ACLF stage, whereas metabolic disruption was significantly downregulated. 16 The relative proportions of innate immune related cells are significantly elevated in monocytes and macrophages, whereas those of adaptive immune-related cells are reduced. 17 Furthermore, in a study involving 40 patients with ACLF in India, 258 plasma metabolites (184 upregulated and 74 downregulated) were differentially expressed, forming distinct clusters in the plasma of patients with ACLF compared with those in the healthy control. 18 The dysregulated metabolites involved in biological processes (BP) in all subtypes were primarily associated with tryptophan, aromatic amino acids, and decreased vitamin and energy metabolism linked to inflammation and immune activation. 18 Therefore, the molecular pathogenesis of ACLF differ significantly among patients with varying ACLF severities; however, the precise molecular mechanisms remain poorly understood.
Experimental mouse models are useful tools for understanding the pathophysiology and molecular mechanisms of human diseases and exploring novel therapeutic strategies. 3 Establishing a stable animal model that mimics the ACLF spectrum is essential to better understand its pathogenesis, prevent the development of life-threatening complications, and improve clinical intervention strategies.3,19 Several ACLF animal models have been established using a combination of chronic and acute liver injuries.3,19,20 Chronic injury is most commonly induced through the intraperitoneal administration of carbon tetrachloride (CCl4) for 10 weeks animal models, whereas, for acute injury, this is followed by injecting a combination of lipopolysaccharide (LPS) and D-galactosamine (D-Gal).19,20 In this study, we developed an ACLF mouse model by chronically administering CCl4 (chronic injury) followed by an acute injection a higher CCl4dose (acute injury). This model closely resembles clinical and pathological features of ACLF, and is therefore a suitable experimental model for investigating the molecular pathogenesis of ACLF.
Comparative proteomic strategies facilitate the study of proteins and their functions, providing insights into the global host protein profile in response to liver injury, and deepening the understanding of the molecular pathogenesis of ACLF. 21 This is supported by the fact that quantitative proteomic analysis has emerged as a powerful technique for identifying protein biomarkers, facilitating the early recognition, diagnostic testing, monitoring, and treatment of various diseases, including ACLF.21–23 Intensive efforts have been made to identify candidate blood biomarkers preferentially expressed in patients with ACLF to facilitate their prospective isolation.24–27 However, investigating molecular differences in the liver tissues of mice with and without ACLF using quantitative proteomics has remained unreported. Therefore, to elucidate the molecular differences influencing the course of liver failure in the development of ACLF, we established an ACLF mouse model and examined the differential expression of proteins in the pooled liver tissues of mice at different disease stages using four-dimensional (4D) label-free liquid chromatography-tandem mass spectrometry (LC-MS/MS). Our results provide new insights into the potential molecular pathogenesis of ACLF development and progression. Moreover, this study provides fundamental information for improving targeted therapeutic intervention strategies, serving as a reference for future in-depth studies to improve the clinical outcomes of patients with ACLF.
Methods
Generation of the animal model and grouping
C57BL/6J mice (male; 8 weeks old; 20–25 g) were provided by the Laboratory Animal Centre of the Affiliated Hospital of Jiaxing University (Jiaxing, China). The mice were maintained under controlled conditions (22 ± 2.5°C, 12 h: 12 h light-dark cycle, and 50% humidity) with free access to standard food and water from March 2023 to September. All laboratory animal handling and experimental procedures were in accordance with the ARRIVE guidelines; and approved by the Institutional guidelines of the Animal Care and Use Committee of Affiliated Hospital of Jiaxing University (Approval ID: JUMC2021-041). Animal discomfort and suffering Efforts were minimized.
Mouse models of liver fibrosis (LF), liver cirrhosis (LC), and ACLF were established as previously described.19,20 Briefly, for the model groups, male C57BL/6J mice were injected intraperitoneally with 20% CCl4 (Sigma-Aldrich, St. Louis, MO, USA) olive solution (2 mL/kg) twice per week for 8 weeks (LF), subsequent injection of a double dose of 20% CCl4 olive solution (4 mL/kg) twice per week for 4 weeks (LC), followed by a combined intraperitoneal injection of 600 µL saline containing 800 mg/kg D-Gal and 10 μg/kg LPS (Sigma-Aldrich, St. Louis, MO, USA) for 6 h (ACLF). The optimal dose for ACLF induction ACLF has been previously described.19,20 In the control (CTRL) group, the mice were administered the same volume of normal olive solution and saline instead of CCl4 or LPS/D-Gal. After 7 days of acclimatization, 100 mice were randomly divided into four groups based on ACLF progression for different periods, namely the control, LF, LC and ACLF following the International Guiding Principles for Biomedical Research Involving Animals (Figure 1). Three biological replicates were analyzed for each group to obtain repeatable accuracy. Flow chart for the generation of carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) treatment (CCl4 + LPS/D-Gal)-induced acute-on-chronic liver failure (ACLF) model mice.
Sample preparation
After establishing the respective mouse models, the mice were anesthetized with 40 mg/kg intraperitoneal sodium pentobarbital, and portal vein blood and fresh liver tissue samples from the respective groups were harvested and immediately frozen in liquid nitrogen after washing with cold phosphate-buffered saline (PBS) within 15 min, and eventually stored at −80°C for further analysis. The inclusion and exclusion criteria for the selection of samples were based on were based on histological examination and biochemical analysis as previously described.3,19,20 The liver samples from the respective groups (n = 18) were thawed, collected and immediately processed following standard operating procedures to minimize preanalytical variation. For each group, six fresh liver tissue samples were mixed to form one biological replicate, and three biological replicates were used to minimize individual differences in the mice for proteomic analysis using label-free quantitative proteomics.
Protein extraction was performed as described previously with minor modifications.23,27,28 Briefly, an appropriate amount of liver tissue sample was weighed, ground into fine powder in liquid nitrogen, and transferred to centrifuge tubes. 4 volumes of lysis buffer (4% sodium dodecyl-sulfate; 100 mM Tris-HCl; pH 7.6) and 1% Protease Inhibitor Cocktail were added, followed by tissue homogenization and sonication using Ultrasonic cell crusher (Scientz 08-IIIC, Ningbo, Zhejiang, China) on ice. After centrifugation at 15,000×g for 12 min at 4°C, the debris was removed, and the protein concentration was determined using the BCA Protein Assay Kit (Bio-Rad, Hercules, USA) following the manufacturer’s protocol.
The filter-aided sample preparation procedure was used for protein digestion with trypsin, as previously described. 23 First, 200 µg of the pooled protein from each sample was sequentially digested using 10 mM dithiothreitol at 37°C for 1.5 h, followed by incubation with 20 mM iodoacetamide at 16°C in the dark for 30 min. After the reaction, the protein samples were transferred to filters and washed with 100 µL UA buffer (8 M urea; 100 mM Tris-HCl; pH 7.6) thrice and then with 100 µL 25 mM NH4HCO3 buffer twice. Subsequently, trypsin (Promega, Madison, WI, USA) was added to the samples at a 1: 50 (trypsin to protein) mass ratio at 37°C for a first 16-h digestion, followed by a second 4-h digestion at a mass ratio of 1:100. Digested peptides were separated through filtration and desalted at room temperature using a C18 Cartridge column (Empore™ SPE Cartridges C18, standard density, bed I.D. 7 mm, volume 3 ml, Sigma-Aldrich, St. Louis, MO, USA). The resultant peptide mixture was further reconstituted in 40 µL of 0.1% (v/v) formic acid after concentration using vacuum centrifugation for the remaining experiments. Peptide content was estimated using UV light spectral density at 280 nm, based on the frequency of tryptophan and tyrosine in vertebrate proteins.
LC-MS/MS analysis
The obtained peptide fractions underwent LC-MS/MS analysis on a timsTOF mass spectrometer (Bruker Daltonics, Bremen, Germany) in a positive ion mode coupled with Evosep One system liquid chromatography (Evosep Biosystems, Odense, Denmark) in a data-dependent acquisition mode for high-energy collision dissociation (HCD) fragmentation. Tryptic peptides were first dissolved in the mobile phase A (0.1% formic acid in water), directly injected into a reverse-phase trap column (Thermo Fisher Scientific), and separated on an EASY-Spray C18-reversed phase analytical column (Thermo Fisher Scientific) using a linear gradient of mobile phase B (0.1% formic acid and 84% acetonitrile) for 120 min at a flow rate of 300 nL/min. Briefly, after nanoflow high-performance liquid chromatography, the most abundant precursor ions were collected from the survey scan ranging from 300 to 1800 m/z with a resolution of 60,000 at m/z 200. The automatic gain control target, dynamic exclusion duration, and maximum ion injection time were set as 1e6, 30 ms and 50 ms, respectively. In tandem mass spectrometry (MS/MS) acquisition, the 20 most abundant precursor ions were chosen to be fragmentized using HCD spectra at a normalized collision energy of 30 eV with a resolution of 15,000 at m/z 200, isolation width of 1.5 m/z, and underfill ratio of 0.1%.
Proteomic data and bioinformatic analysis
Following peptide separation, the resulting raw mass spectrometry data for each sample were combined and searched using the MASCOT database (Matrix Science, London, UK, version 2.5.1) of the Proteome Discoverer 1.6 software against a uniport mouse database provided by the Universal Protein Resource (UniProt; https://www.uniprot.org, released on 2023-10-11, with 27,669 entries). For peptide identification and quantification, searches were performed using the following parameters: enzyme, trypsin; fixed modification, carbamidomethyl (C); maximum missed cleavage, two; variable modification, oxidation (M); main search, 6 ppm; first search and MS/MS tolerance, 20 ppm; database pattern, reverse; protein quantification, intensity-based absolute and label-free quantification. Additionally, all significant quantitative data were reported at a 99% confidence level for protein and peptide identification, as determined using a false discovery rate (FDR) threshold of 1.0.
Significantly differentially expressed proteins used for further analysis were those with a fold change ≥2 or ≤0.5 with p < 0.05, as determined using a paired t-test. The significantly differentially expressed proteins underwent hierarchical clustering analysis using Cluster 3.0 software (available at https://bonsai.hgc.jp/∼mdehoon/software/cluster/software.htm) and Java TreeView software (available at https://jtreeview.sourceforge.net). Enrichment analysis was conducted using Fisher’s exact test, with the entire set of quantified proteins serving as the background dataset. To account for multiple testing, the Benjamini-Hochberg correction was applied to adjust the resulting p-values. Only functional categories and pathways with p-values below the threshold of 0.05 were deemed significant. The CELLO system (https://cello.life.nctu.edu.tw/), a multi-class support vector machine (SVM) classification system, was employed to predict protein subcellular localization. Gene ontology (GO) annotation and pathway enrichment analysis of the differentially expressed proteins were conducted using Blast2GO software with default options based on functional annotations related to BP, molecular functions (MF), and cellular components (CC). Subsequently, the selected differentially expressed proteins were searched against the online Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://geneontology.org/) to annotate their KEGG orthology identities and the protein pathways were searched against the KEGG Automatic Annotation Server (https://www.genome.jp/tools/kaas/). The protein–protein interaction (PPI) data for the proteins under investigation were obtained from STRING software (https://string-db.org/). The resulting data were downloaded in XGMML format and subsequently imported into Cytoscape software (https://www.cytoscape.org/, version 3.2.1) for visualization and further analysis of the functional PPI networks. Additionally, the degree of connectivity for each protein was computed to assess its significance within the PPI network.
Validation of selected dysregulated proteins using immunohistochemistry (IHC) and quantitative real-time polymerase chain reaction (qRT-PCR)
To confirm the proteomic findings of this study, proteins exhibiting substantially different levels in the four groups underwent IHC and qRT-PCR, as previously reported.29,30 IHC was performed following the manufacturer’s instructions. Briefly, liver tissue samples were fixed and embedded in 4% paraformaldehyde. After routine deparaffinization in xylene and gradient dehydration using ethanol, the paraffin sections were covered with peracetic acid, sent for antigen retrieval, and incubated with the primary antibody at 4°C overnight. Subsequently, the sections were submerged in the corresponding biotinylated secondary antibody at 16°C for another 1 h and visualized using 3, 3′-diaminobenzidine. The final intensity score was classified as 0 (negative staining), 1 (minimal staining), 2 (moderate staining) or 3 (strong staining).
For qRT-PCR, total RNA was extracted from 72 fresh frozen liver tissue samples using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and quantified using a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA) following the manufacturer’s instruction. The expression of myeloperoxidase (MPO) and coronin-1A (CORO1A) RNA was analyzed through quantitative reverse transcription-PCR using SYBR Green PCR Master Mix (Life Technologies, Carlsbad, CA, USA) on a Step One Plus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The sequences of the gene-specific primers used to detect the expression of these genes were designed using the Primer-Blast Database. The 5′ to 3′ primer sequences were as follows: MPO-forward primer, GACATGCCCACCGAATGACAA; MPO-reverse primer, CAGGCAACCAGCGTACAAAG; CORO1A-forward primer, CACCCGGACACAATCTACAGC; CORO1A-reverse primer, CTCGATGACACGAACTCGCTT; β-actin-forward primer, ATAGCACAGCCTGGATAGCAACGTAC; β-actin-reverse primer, CACCTTCTACAATGAGCTGCGTGTG. After qRT-PCR, the relative mRNA levels of each cellular target were normalized to the geometric mean of β-actin and calculated by comparing the threshold cycle values.
Statistical analysis
Quantitative data obtained from at least three independent replicates and data were processed and analyzed using Student’s t-test to compare between the two groups and expressed as the mean ± standard deviation. Statistical significance was set at p < 0.05. Correlations between variables were assessed using Spearman’s rank correlation test. Plotting and statistical analyses were conducted using the SPSS software (version 23.0) package and GraphPad Prism software (version 9.0), respectively.
Results
Combinations of LF, LC, and LPS/D-Gal-induced ACLF in mice
Based on the clinical course and pathological mechanisms of ACLF, we first developed an ACLF animal model by inducing LF and LC as chronic liver injuries. We combined the following treatments to simulate the three major stages: CCl4 administration 8 weeks (chronic, LF), subsequent injection of a double CCl4 dose for 4 weeks (LC), and a combined intraperitoneal injection of D-Gal/LPS for 6 h (ACLF). Our results show 7 out of 25 mice died in the ACLF group. This high mortality rate is similar to that previously described in other studies of ACLF patients.6,7 Compared with that of the control, LF, and LC groups, liver morphological changes in the ACLF group were significantly different (Figure 2(a) and (b)). This was demonstrated by the destruction of hepatic architecture, widespread hepatocyte necrosis, extensive infiltration of inflammatory cells around the central veins and bleeding spot dissemination. Additionally, some serum indicators of the extent of liver tissue damage displayed significant differences among the four groups. Levels of alanine transaminase (10,241.67 ± 1083.03 U/L vs 133.67 ± 18.02 and 619.00 ± 174.76 U/L), aspartate transaminase (10,683.83 ± 1512.54 U/L vs 710.50 ± 129.31 and 955.67 ± 149.07 U/L) and total bilirubin (67.50 ± 13.63 μmol/L vs 35.00 ± 8.99 and 55.67 ± 12.11 μmol/L) were significantly higher in the ACLF group than in the LF and LC groups (Figure 2(c)). Therefore, the ACLF mouse model induced by CCl4 + LPS/D-Gal most closely replicates ACLF features, providing a reliable alternative for investigating ACLF molecular mechanisms and exploring potential novel therapeutic strategies. Effect of carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) treatment (CCl4 + LPS/D-Gal) on liver hepatic pathology. (a) Photograph of liver tissue. (b) Representative photomicrographs of haematoxylin and -eosin (H&E) staining. (c) Serum alanine transaminase (ALT), aspartate transaminase (AST), and total bilirubin (TBIL) levels in mice with CCl4 + LPS/D-Gal treatment for different periods.
Relative quantification of the liver proteome associated with ACLF in mice
Mouse tissue samples were analyzed for global profiling of protein expression using a high-throughput label-free LC-MS/MS method (Figure 3(a)). After searching the mouse UniProt proteome database using the integrated Andromeda search engine, we identified 4226 unique proteins across 26,743 matched peptides resulting from 202,618 matched spectra (Figure 3(b)). Among these, 4,057, 4,196, 4,147, and 4201 proteins were quantified with three biological replicates in the CTRL, LF, LC, and ACLF groups, respectively, and 4007 overlapping proteins were shared among the four groups, accounting for 94.82% of the total identified proteins (Figure 3(c)). Additionally, the number of proteins based on the Andromeda score (Figure S1a), molecular weight (Figure S1b), peptide count (Figure S1c), and peptide coverage distribution (Figure S1d), a peptide scores based on mass error distribution (Figure S1e) indicated that the overall proteome datasets of the four liver tissue samples had no strong bias. This suggests that the proteomic data met acceptable requirements and had high reliability. Features of the proteome data set or liver tissue samples from carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) treatment (CCl4 + LPS/D-Gal)-induced acute-on-chronic liver failure (ACLF) mouse models. (a) overall technical route of this project. (b) The number of spectra, peptides and proteins. (c) Venn diagrams showing the number of identified proteins and the protein identification overlaps in the four groups.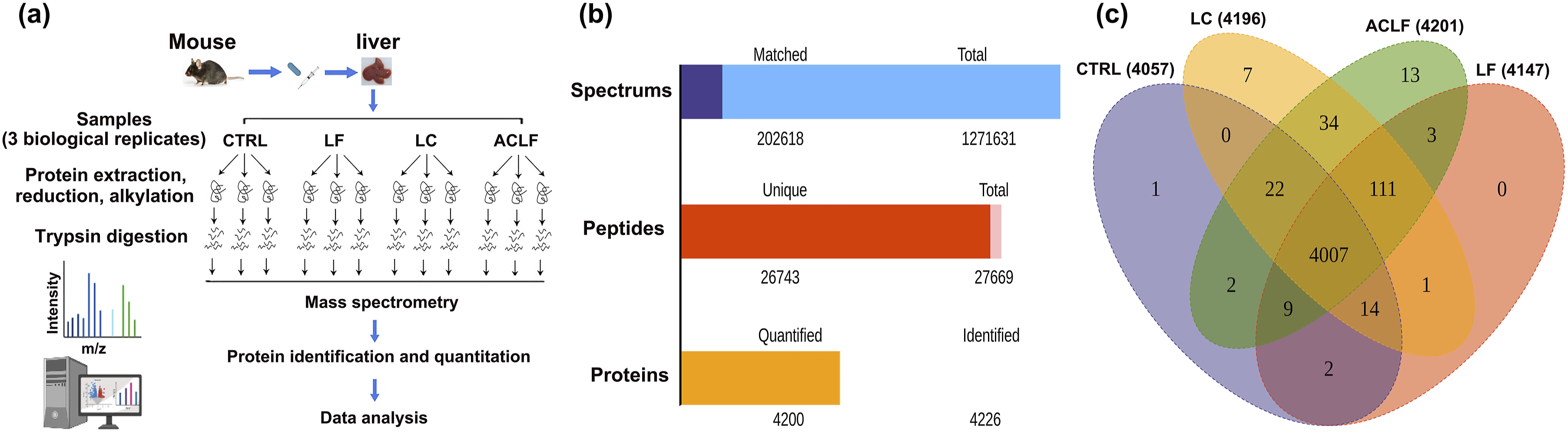
Identification of the differentially expressed proteins associated with ACLF
To identify the differentially expressed proteins, we compared the relative protein expression values between the selected groups (LF vs CTRL, LC vs CTRL, and ACLF vs CTRL). Compared with those in the control group, 258 (162 upregulated and 96 downregulated) (Figure 4(a) and (b); Table S1) and 380 (253 upregulated and 127 downregulated) (Figure 4(d) and (e); Table S2) proteins with a mean expression fold change ≥2 and p < 0.05 were differentially expressed in the LF and LC groups, respectively. The typically differentially expressed proteins are listed in Table 1. Moreover, these protein sets were clearly separated into distinct clusters, as expected. Dysregulated proteins underwent GO enrichment analysis and were categorized according to BP, MF, and CC to elucidate their functions and features. In the BP category, the top enriched terms were cellular processes, metabolic processes, and biological regulation, whereas the proteins with differential abundance were primarily involved in binding and catalytic activity in both groups (Figure 4(c) and (f)). Bioinformatics analysis of differentially expressed proteins from the liver samples of the liver fibrosis (LF) and liver cirrhosis (LC) mouse groups compared with that of the control (CTRL) mouse group. (a) Volcano plot representing protein abundance changes (LF vs CTRL). A total of 258 dysregulated proteins with fold change ≥2 and p-values <0.05 were identified. (b) Hierarchical clustering analysis of the 258 dysregulated proteins (LF vs CTRL). (c) Gene ontology (GO) analysis of 258 dysregulated proteins (LF vs CTRL). (d) Volcano plot representing protein abundance changes (LF vs CTRL). A total of 380 dysregulated proteins with fold change ≥2 and p-values <0.05 were identified. (e) Hierarchical clustering analysis of the 380 dysregulated proteins (LF vs CTRL). (f) GO analysis of 380 dysregulated proteins (LF vs CTRL). List of the typically differentially expressed proteins of liver tissue samples from carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) treatment (CCl4 + LPS/D-Gal)-induced acute-on-chronic liver failure (ACLF) mouse models.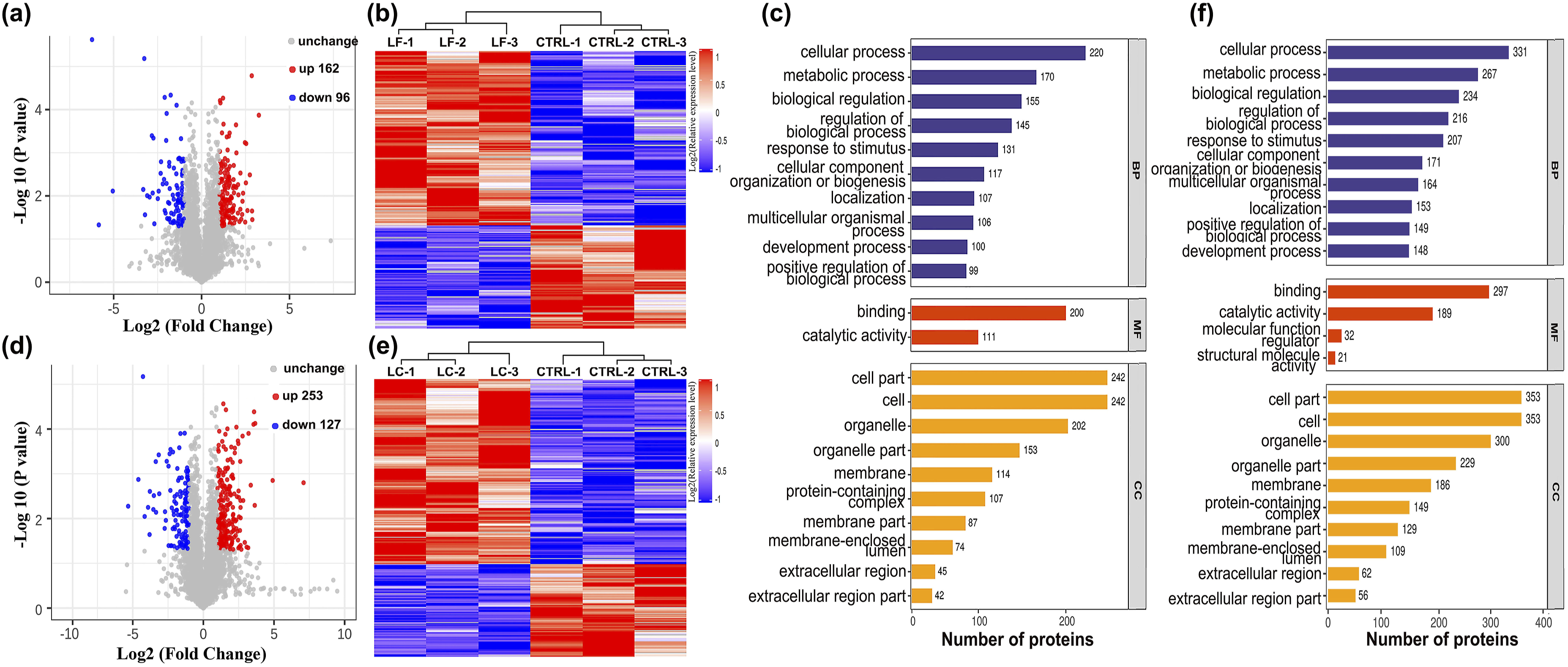

Similarly, liver samples between the ACLF and control groups were compared using the aforementioned criteria. In the hierarchical clustering analysis, 471 proteins (319 upregulated and 152 downregulated) were differentially expressed, forming distinct clusters in the livers of ACLF model mice compared with those in the livers of normal controls (Figure 5(a) and (b); Table S3). GO enrichment analysis of three distinctive function sets related that, in the BP category, 408 proteins were involved in the cellular processes, 313 proteins in metabolic processes, and 303 proteins in biological regulation. In the MF category, the two most frequently occurring terms were binding (360 proteins) and catalytic activity (203 proteins), followed by MF regulation (43 proteins); cell part (439 proteins), cell (439 proteins), and organelle (374 proteins) were the top three ranked terms in CC (Figure 5(c)). Subsequently, we investigated subcellular localization with at least one term assigned to each of the identified proteins using GO annotation and found that over 51.59% of the proteins were annotated as cytoplasmic, and the other three primary categories of these dysregulated proteins were nuclear (154 proteins), cell membrane (87 proteins), and secreted (73 proteins) (Figure 6(d)). Although cytoplasmic, nuclear, and cell membrane proteins were the most abundant categories, multi-pass membrane proteins, endoplasmic reticulum membranes, and cytoskeleton proteins were also readily identified. Therefore, our findings suggest that many BPs, MFs and CCs are extensively affected in mice with ACLF and may have different molecular backgrounds and mechanisms compared with those in the control animals. Bioinformatics analysis of differentially expressed proteins between the liver samples of the acute-on-chronic liver failure (ACLF) and the control (CTRL) mouse groups. (a) Volcano plot representing protein abundance changes (ACLF vs CTRL). A total of 471 dysregulated proteins with fold change ≥2 and p-values <0.05 were identified. (b) Hierarchical clustering analysis of 471 dysregulated proteins (ACLF vs CTRL). (c) Gene ontology (GO) analysis of 471 dysregulated proteins (ACLF vs CTRL). (d) Subcellular localization of 471 dysregulated proteins. Kyoto Encyclopaedia of Genes and Genomes (KEGG) annotation of the dysregulated proteins involved in carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) treatment (CCl4 + LPS/D-Gal)-induced acute-on-chronic liver failure (ACLF) mouse liver samples. (a) KEGG pathway-based enrichment analysis of 258 dysregulated proteins (liver fibrosis [LF] vs control [CTRL]). (b) KEGG pathway-based enrichment analysis of 380 dysregulated proteins (liver cirrhosis [LC] vs CTRL). (c) KEGG pathway-based enrichment analysis of 471 dysregulated proteins (ACLF vs CTRL). (d) Predicted network of the dysregulated proteins between ACLF and CTRL group liver samples.

Furthermore, the protein expression alternation in liver samples between the ACLF and LC groups was also analyzed using the aforementioned criteria, and 103 dysregulated proteins (Table S4 and Supplemental Figure S2), including 66 upregulated and 39 downregulated proteins were found (Figure S2a). The upregulated and downregulated proteins formed distinct clusters (Figure S2b), and most of these dysregulated proteins have been reported to be closely, associated with different biological processes, including cellular processes, metabolic processes and biological regulation (Figure S2c and S2d). These results can clearly indicate different molecular backgrounds and molecular mechanisms between the ACLF and LC mouse groups.
KEGG pathway analysis of dysregulated proteins
To further understand the roles of proteins with ACLF-associated expression alterations, we conducted a KEGG pathway-based enrichment analysis of the dysregulated proteins, which were subsequently mapped to reference KEGG pathways. Between the LF and control groups, the dysregulated proteins were primarily enriched in pathways related to Alzheimer’s disease, neurodegenerative diseases, amyotrophic lateral sclerosis, Salmonella infection, phagosomes, and cancer (Figure 6(a)). Between the LC and control groups, the dysregulated proteins were primarily enriched in pathways related to human papillomavirus infection, neurodegenerative diseases, pathways in cancer, Alzheimer’s disease, focal adhesion, and phagosome (Figure 6(b)). Furthermore, comparative analysis of liver tissues from the ACLF model and normal control mice; revealed that the pathways were enriched with proteins significantly linked to neurodegenerative diseases, signal transduction, transport and catabolism, and cancer. Similarly, these pathways exhibited focus hubs containing pathways related to neurodegeneration, including multiple diseases (24 proteins), amyotrophic lateral sclerosis (22 proteins), PI3K-Akt (20 proteins), Alzheimer’s disease (19 proteins), phagosomes (18 proteins), and cancer (18 proteins), which were the top six regulated signaling pathways enriched with differentially expressed proteins (Figure 6(c)). Thus, the results revealed that specific signaling pathways were differentially modulated in mice with ACLF, although common signaling pathways were identified. Among these categories, the phagosome pathway was the top modulated signaling pathway for enriching dysregulated proteins, which were at the core of the protein-protein interaction (PPI) network, suggesting that they may have contributed to the regulation of clinical features and a relatively vital biological function in ACLF pathogenesis (Figure 6(d) and Figure 7). Therefore, the phagosome pathway may contribute to the regulation of immune-mediated liver tissue damage, massive hepatocyte necrosis, and liver failure during the development of ACLF through the regulation of dysregulated proteins. Phagosome pathway obtained from the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway-based enrichment analysis of carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) treatment (CCl4 + LPS/D-Gal)-induced acute-on-chronic liver failure (ACLF) mouse liver samples.
Verification of the differentially expression of MPO and CORO1A
According to the key modulated signaling pathways (Figure 7) and hierarchical clustering analysis results (Figure 5), the proteins MPO (51.67-fold) and CORO1A (6.31-fold) were differentially expressed between ACLF and normal control mice. Therefore, they may potentially induce ACLF in CCl4 + LPS/D-Gal-treated mice by regulating the phagosome pathway. Hence, to further confirm the quantitative proteomic results, we investigated changes in MPO and CORO1A expression in normal control, LF, LC, and ACLF mouse liver tissues using IHC analysis. The MPO and CORO1A expression levels were increased gradually from the early to late stages of ACLF development in 72 paraffin-embedded liver specimens and correlated with the extent of liver tissue damage (Figure 8(a)). Validation of the selected differentially expressed proteins myeloperoxidase (MPO) and coronin-1A (CORO1A) from the liver tissues of the liver fibrosis (LF), liver cirrhosis (LC), and acute-on-chronic liver failure (ACLF) mouse groups compared with that in the control (CTRL) mouse group. (a) Representative image of MPO and CORO1A immunohistochemical staining in mice with carbon tetrachloride (CCl4) coupled with lipopolysaccharide (LPS)/D-galactosamine (D-Gal) administration (CCl4 + LPS/D-Gal) for different periods, respectively. (b) Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of MPO and CORO1A mRNA expression levels in mice with CCl4 + LPS/D-Gal treatment for different periods, respectively. *p < 0.05, **p < 0.01.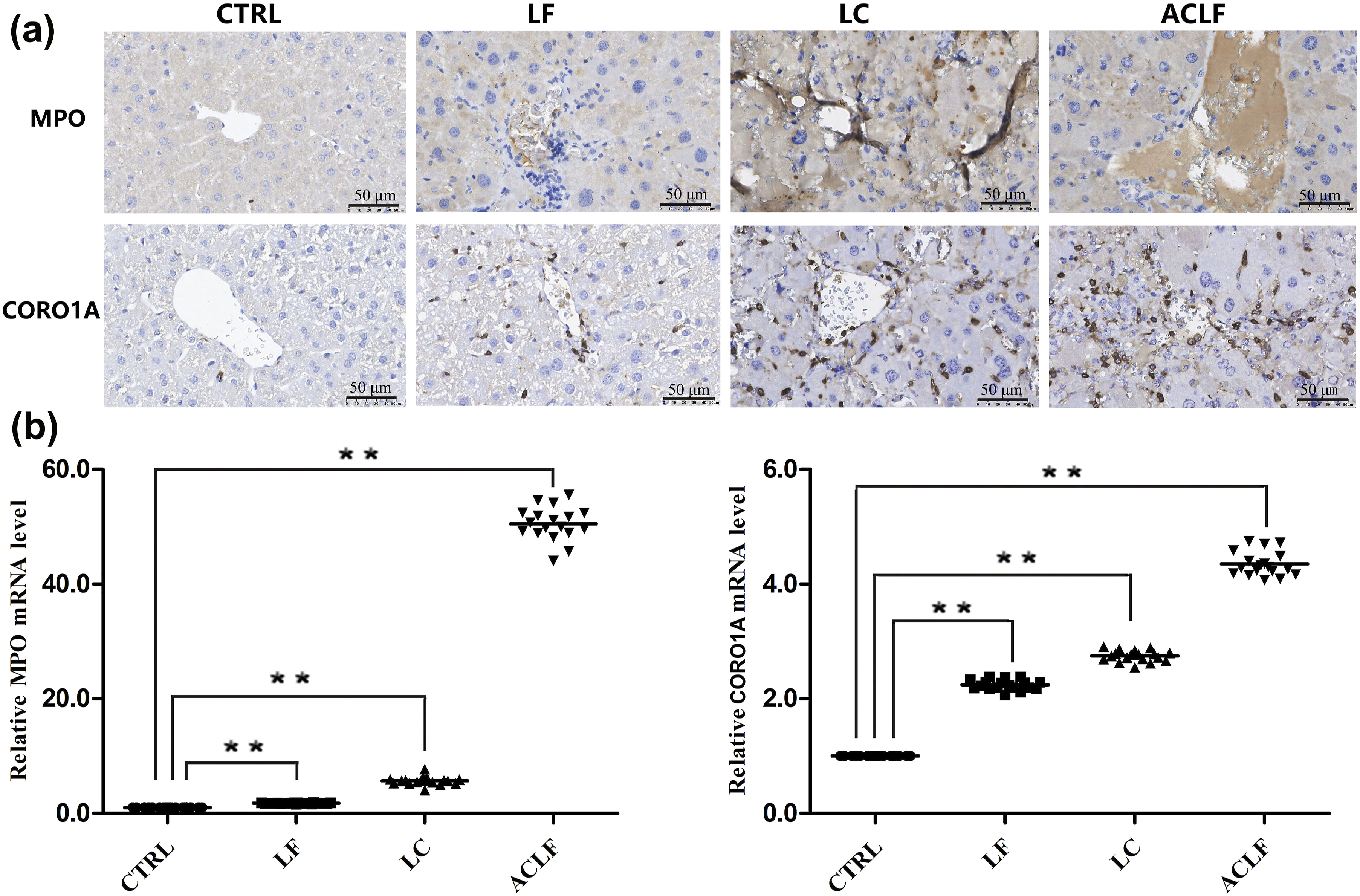
qRT-PCR was performed to validate the relative MPO and CORO1A mRNA expression levels. MPO mRNA expression levels were significantly upregulated by 50.52-fold (n = 18 mice), whereas the CORO1A mRNA expression levels were upregulated by 5.35-fold (n = 18 mice) in ACLF mouse liver samples compared with those in the control samples (Figure 8(b)). Consistent with those of LC-MS/MS, the IHC and qRT-PCR results of liver tissue sections further confirmed the increased of MPO and CORO1A from mice treated with CCl4 + LPS/D-Gal compared with control group. Thus, given the consistency and comparability of the analytical results, MPO and CORO1A may play crucial roles in inducing ACLF in CCl4 + LPS/D-Gal-treated mice by regulating the phagosome pathway. However, the underlying molecular mechanisms require further investigation.
Discussion
Despite considerable advances in understanding and treating ACLF, it remains a significant global health concern because of its association with high short-term morbidity, which worse than that of acute liver failure (ALF).1,31 Patients with ACLF constitute a major proportion of fatal hepatic cirrhosis cases, and ACLF is associated with a life-threatening syndrome accompanied by one or more extrahepatic organ failures and an increased risk of short-term mortality.13,32,33 Previous studies have focused on the disparities in clinical characteristics between patients with ACLF and healthy individuals, with the underlying mechanism and alterations in biological pathways involved in ACLF development largely unexplored.33,34 Therefore, conducting a liver proteomic analysis to identify factors distinguishing ACLF liver tissue from normal liver tissue is a crucial for evaluating the proteins and cellular pathways that regulate ACLF progression. Notably, the liver proteomic data presented in this study are the first to revel molecular differences among LF, LC, ALF, and CTRL mice at the proteome level. These data confirm those of previous studies by revealing that an ALF model induced using by CCl4 + LPS/D-Gal could be used to investigate ACLF development. Moreover, this study provides a theoretical basis for the molecular mechanism underlying organ dysfunction or failure related to the pathogenesis and progression of ACLF, which will facilitate further comprehensive studies.
The clinical course and pathological mechanism of ACLF can be divided into three primary stages, namely LF, LC, and ACLF.6,33 Although several experimental ACLF animal models have been reported, none has recapitulated or simulated the entire pathological process of this disease.3,19,20 In this study, we confirmed that the CCl4 + LPS/D-Gal-induced ACLF mouse model was the closest to human ACLF and may be a reliable alternative for studying the molecular mechanisms of ACLF (Figure 2). To obtain comprehensive insights into the molecular pathogenesis of ACLF and identify new therapeutic targets, we systematically investigated changes in the liver proteome. We used label-free LC-MS/MS-based quantitative proteomic analysis of liver tissue samples from four groups (CTRL, LF, LC, and ACLF) to profile dynamic changes in the global liver proteome. Among the 4007 overlapping proteins with FDR <1%, 94.82% were common to all four groups, indicating the stability of the workflow and reliability of the study conclusions (Figure 2(c)). Based on the identification criteria for differentially expressed proteins (fold change ≥2; p < 0.05), 258, 380, and 471 dysregulated proteins were identified in the liver samples of LF, LC, and ACLF mouse groups, respectively, compared with those in the normal control. Alterations in the expression of the identified proteins in different mouse models involved different BP, including cellular processes, metabolic processes, biological regulation, BP regulation, response to stimuli, and localization, confirming that the pathogenesis of LF, LC, and ACLF is associated with different molecular mechanisms. Notably, most identified dysregulated proteins (ACLF vs CTRL) were associated with binding, catalytic activity, MF regulation, structural molecule activity, and transporter activity, which primarily reflect organ tissue injury and metabolic disorders. For example, patients with ACLF have a significant higher risk of vital organ injury and metabolic disorders and poorer clinical outcomes than those without ACLF.4,14,31 Therefore, individuals with organ injury and metabolic disorders have a higher incidence of ACLF progression than healthy individuals do. This is further supported by the altered signaling pathways in the liver tissues of ACLF and normal control mice, which are linked to neurodegenerative disorders, signal transduction, transport, and catabolism during ACLF development. Although the signaling pathways involved with the aforementioned differentially expressed proteins were vital in ACLF development, the phagosome pathway was consistently implicated in the comparison groups, and the enrichment of dysregulated proteins was primarily at the core of the PPI network. Hence, these data strongly support our initial hypothesis that ACLF progression may have different pathological mechanisms and that the phagosome signaling pathway may contribute to the regulation of ACLF clinical features and progression and may be used as a novel candidate target for ACLF treatment.
The phagosome is a vesicle that forms around particles ≥0.5 μm in diameter by fusing with cell membranes containing pathogens and particulates. This vesicle is a crucial organelle for the innate ability of macrophages to participate in tissue remodelling, clear apoptotic cells, and restrict the spread of intracellular pathogens through a phagocyte via phagocytosis.35,36 Phagosomes undergo a complex maturation process with vesicles in the endolysosomal system, creating an increasingly acidic and hydrolytic environment within the phagosomal lumen. This process is termed phagolysosome biogenesis.37,38 This pathway involves several interactions with other intracellular organelles, including recycling endosomes, late endosomes, and lysosomes, facilitating vacuolar-type adenosine triphosphatase-dependent acidification, regulation of metal ion fluxes, and generation of reactive oxygen species, nitrogen species, and hydrolytic enzymes, involved in the killing and degradation of phagosome content.37,38 Hence, phagosomes play an essential role in activating inflammatory and immune responses against invading pathogens. Moreover, they play a vital role in cellular homeostasis by removing senescent and apoptotic cells and cellular or foreign debris through dedicated phagocytes such as dendritic cells, monocytes, macrophages, and neutrophils.35,38–40 In CCl4 + LPS/D-Gal treatment, activated phagocytosis involves activating inflammatory and immune responses, maintaining cellular homeostasis, and determining the magnitude of liver injury severity and disease pathogenesis. Thus, our data support the hypothesis that the degree of activation or inhibition of the phagosome signaling pathway determines the effects of phagocytosis on the hepatic manifestation of CCl4 + LPS/D-Gal-induced ACLF in mice. Therefore, the phagosome signaling pathway is a critical component of the immune defense and liver homeostasis mechanisms, acting an important role in the pathogenesis of liver damage associated with CCl4 + LPS/D-Gal-induced ACLF.
Furthermore, our analysis also revealed that the proteins MPO and CORO1A were the most upregulated in liver samples from the ACLF mouse group compared with those in liver samples from the CTRL, LF, and LC groups. MPO (also known as verdoperoxidase) is a member of the heme peroxidase cyclooxygenase enzyme family and is primarily synthesized in neutrophils and, to a lesser degree, in monocytes.41,42 The protein plays crucial roles in innate immune defense, inflammatory responses, heart failure, obesity, hypertension, and insulin resistance by facilitating the formation of reactive oxygen species in the extracellular space or within the phagosomes to destroy engulfed microorganisms utilizing H2O2 and a halide or pseudohalide.41–43 We previously demonstrated that neutrophil-derived MPO is a crucial factor in liver tissue defense, and MPO oxidants play a significant proinflammatory role by promoting host tissue damage and activating the NETosis signaling pathway during LPS/D-Gal-induced ALF. 22 Here, activation of phagocytosis upregulated MPO expression, increasing its affinity for damaged endoplasmic reticulum-mediated phagocytic components and causing exacerbated liver inflammation, disrupted tissue homeostasis, and dysfunction in a murine model of CCl4 + LPS/D-Gal-induced ACLF. This suggests that MPO-damaged biomolecules and fragments are associated with poor liver function, and can modulate ACLF progression.
CORO1A (TACO or p57) is, a member of the coronin I protein family and is abundantly expressed in the cytosol and cell cortex. The protein displays a complex structure that includes an N-terminal region with seven tryptophan-aspartic repeats and a C-terminal coiled coil, playing a pivotal role in T lymphocyte trafficking, cell migration and division, vesicle transport, and calcium signal transduction.44–47 CORO1A is required for regulating actin cytoskeleton disassembly and cytoskeleton-dependent processes, including phagocytosis.48,49 For example, during signal transduction, CORO1A is localized in mycobacterial phagosomes, functioning as a phagosomal coat protein that promotes the survival of M. tuberculosis in phagocytic cells in pathogen–host interactions. 50 In this study, upregulation of CORO1A expression increased susceptibility to phagocytosis-mediated liver damage, fibrosis, and cirrhosis, which are crucial in ACLF progression. Consistent with the KEGG pathway analysis, which revealed that the phagosome pathway involved in inflammation and tissue homeostasis was a vital pathogenic mechanism in ACLF; several functionally associated proteins, including MPO and CORO1A, were dysregulated. The phagosome pathway is crucial in the development of CCl4 + LPS/D-Gal-induced ACLF by regulating the expression of several proteins, including MPO and CORO1A. In this study, compared with that of the control, LF, and LC groups, liver morphological changes and hepatocyte injury caused by CCl4 + LPS/D-Gal in the ACLF group were significantly different and showed more inflammatory cell recruitment (Figure 2(a) and (b)); serum analysis showed increased ALT, AST, and TBiL levels in mice injected with CCl4 + LPS/D-Gal in comparison to normal control group (Figure 2(c)). Furthermore, MPO and CORO1A upregulation activated the phagosome pathway in CCl4 + LPS/D-Gal-induced ACLF mouse liver samples compared with that in normal control mouse liver samples, suggesting that lymphocyte and neutrophils are correlated with the degree of liver damage and may be important prognostic determinants in patients with ACLF. However, the molecular mechanisms by, which MPO and CORO1A activate the phagosomal pathway remain unclear.
We used a 4D-label-free-based quantitative proteomic approach to profile liver proteins from CTRL, LF, LC, and ACLF mouse models. The phagosome pathway was identified as a target for preventing and treating ACLF disease and related tissue damage by regulating MPO and CORO1A. However, the limitations of this study include the small sample sizes, other measures of organ functionality, and collagen stain analysis of liver tissues. Furthermore, we tested only a single concentration of CCl4 + LPS/D-Gal and did not use various doses for parallel comparisons; hence, we did not evaluate whether the effects were drug concentration-dependent. Additionally, these data provide only associative evidence for the role of the phagosome pathway in CCl4 + LPS/D-Gal-induced ACLF; however, the underlying mechanisms by, which MPO and CORO1A activate the phagosomal pathway remain poorly understood. Next, we will investigate the validity and role of crucial differentially expressed proteins through biochemical analyses using stable and reliable experimental ACLF animal models with MPO and CORO1A knock-out mice to understand the molecular pathogenesis of ACLF.
Conclusion
In this study, we used 4D-label-free-based quantitative proteomic analysis to investigate alterations in the liver proteome profile of CTRL, LF, LC, and ACLF mice. The results confirmed our hypothesis regarding the involvement of different protein profiles and related signaling pathways associated with ACLF development. These altered proteins contribute to the development of CCl4 + LPS/D-Gal-induced ACLF by regulating the activation of various signaling pathways, including the phagosome pathway. Although quantitative proteomic analysis remains mostly descriptive, the results of this study provide mechanistic insights into CCl4 + LPS/D-Gal-induced ACLF. Moreover, the study reveals the underlying application of phagosome pathway-associated dysregulated proteins, such as MPO and CORO1A, as candidate targets and/or critical predictors for the prognosis and treatment of ACLF.
Supplemental Material
Supplemental Material - Comparative liver proteomic analysis of protein expression changes in an acute-on-chronic liver failure mouse model
Supplemental Material for Comparative liver proteomic analysis of protein expression changes in an acute-on-chronic liver failure mouse model by Keyin Wang, Zhuolin Zou and Dahai Wei in European Journal of Inflammation
Supplemental Material
Supplemental Material - Comparative liver proteomic analysis of protein expression changes in an acute-on-chronic liver failure mouse model
Supplemental Material for Comparative liver proteomic analysis of protein expression changes in an acute-on-chronic liver failure mouse model by Keyin Wang, Zhuolin Zou and Dahai Wei in European Journal of Inflammation
Supplemental Material
Supplemental Material - Comparative liver proteomic analysis of protein expression changes in an acute-on-chronic liver failure mouse model
Supplemental Material for Comparative liver proteomic analysis of protein expression changes in an acute-on-chronic liver failure mouse model by Keyin Wang, Zhuolin Zou and Dahai Wei in European Journal of Inflammation
Footnotes
Acknowledgements
The authors would like to thank Applied Protein Technology (Shanghai, China) Co., Ltd. for technical assistance on mass spectrometry.
Ethical considerations
Ethical approval for this study was obtained from * Affiliated Hospital of Jiaxing University OF ETHICS COMMITTEE OR INSTITUTIONAL REVIEW BOARD (JUMC2021-041)*.
Author contributions
Dahai Wei and Zhuolin Zou made substantial contributions to conceive and design of the experiments. Keyin Wang performed most of the investigations and experiments. Dahai Wei drafted and edited this manuscript. All authors reviewed and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Zhejiang Provincial Medical Scientific Research Foundation of China under Grant No. 2025KY345 and 2025KY346, Zhejiang Provincial Natural Science Foundation of China under Grant No. LTGY24C010001, Jiaxing Key Laboratory of Virus-related Infectious Disease, and 2023 Jiaxing Key Discipline of Medcine-Lemology (Supporting Subject, Grant No. 2023-ZC-009).
Declaration of conflicting interests
The authors declared no potential conflicts of interests with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Animal welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.