
Abstract
Background
The use of dimethyl fumarate has not been reported in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis.
Objectives
The purpose of this study was to evaluate the efficacy and safety of dimethyl fumarate in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis.
Methods
APEX was a phase 3, multinational trial, which consisted of a 24-week, randomized (1:1), double-blind study where patients received dimethyl fumarate 240 mg or placebo twice daily, followed by an open-label extension where all patients received dimethyl fumarate 240 mg. The primary endpoints were the total number of new gadolinium-enhancing (Gd+) lesions in Weeks 12–24 (Part I) and long-term safety (Part II). This post-hoc subgroup analysis evaluated the efficacy and safety of dimethyl fumarate in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis (n=52) up to Week 72 (24 weeks Part I and 48 weeks Part II).
Results
Dimethyl fumarate reduced the mean total number of new gadolinium-enhancing lesions at Weeks 12–24 by 94% versus placebo; the number of patients who had a relapse over 24 weeks was reduced by 72%. Adverse events leading to discontinuation of the study drug were reported in 9% of patients receiving placebo/dimethyl fumarate and 4% of patients in dimethyl fumarate/dimethyl fumarate.
Conclusions
Dimethyl fumarate demonstrated sustained efficacy and acceptable tolerability in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis for 72 weeks.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disorder of the central nervous system (CNS) with an estimated prevalence of 30 per 100,000 persons. 1 MS is characterized by auto-immune lymphocytic infiltration of the blood-brain barrier and oxidative stress causing demyelination.2,3 These adjustments manifest as episodes of neurological dysfunction, followed by recovery. Relapsing–remitting MS (RRMS) accounts for approximately 80–85% of MS cases at diagnosis. 4 In the later stages of MS, extensive neuronal loss and gliosis are evident resulting in neurodegeneration and reduced recovery. 5 Therefore, the initiation of disease-modifying drugs (DMDs) early in the disease course could slow the accumulation of neurological damage and may contribute to clinically significant results in delayed disease progression. 6
In Japan, DMDs currently available include subcutaneous or intramuscularly administered self-injections of interferon (IFN)-β and glatiramer acetate, intravenous natalizumab, and oral fingolimod and dimethyl fumarate (DMF). DMF has anti-inflammatory, cytoprotective and immunomodulating properties that appear to be mediated through activation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and the hydroxycarboxylic acid receptor 2 (HCAR2).7,8 DMF has been approved in the USA and EU for the treatment of RRMS, including use as first-line therapy; 9 approval in Japan was granted for MS in December 2016. 10 Two phase 3, double-blind, placebo-controlled, multinational studies (DEFINE and CONFIRM), have assessed the efficacy and safety of DMF compared with placebo; individual results and the corresponding integrated analysis of these studies demonstrated a significant reduction in relapse rates and improved measures of disease progression, including the number of gadolinium-enhancing (Gd+) and T2 hyperintense lesions with DMF compared with placebo.11–13 Furthermore, in a five-year interim analysis of ENDORSE (a long-term extension of DEFINE/CONFIRM), DMF demonstrated an acceptable safety profile and the incidence of adverse events (AEs), serious AEs, and discontinuations due to AEs in patients who continued DMF was similar among those receiving DMF/DMF and placebo/DMF. 14
This interim subgroup analysis aimed to evaluate the efficacy and safety of DMF for up to 72 weeks in treatment-naïve Japanese patients with RRMS enrolled in the APEX study which consisted of an initial 24-week randomized, double-blind and placebo-controlled trial (Part I) and an open-label extension (Part II).
Materials and methods
Study design and participants
APEX (ClinicalTrials.gov record: NCT01838668) was a phase 3, two-part study, conducted in 54 locations in Japan, South Korea, Taiwan, Czech Republic and Poland. Patients in Part I were randomized (1:1) to receive 24 weeks of either oral DMF 240 mg or placebo twice daily. Treatment was titrated in Part I of this study, with patients initiating treatment with one capsule (DMF 120 mg or placebo) twice daily. After eight days, patients received two capsules twice daily. Part II was an open-label extension phase to determine the safety profile of DMF in patients with RRMS who completed Part I (Supplemental Material Figure S1), the cut-off date for this interim analysis was 29 April 2016. Patients who received DMF in Part I followed by DMF in Part II were classified as DMF/DMF; those who received placebo in Part I and DMF in Part II were known as placebo/DMF.
Part I of the study included patients aged 18–55 years old with a diagnosis of RRMS according to the revised McDonald criteria, 16 who had an Expanded Disability Status Scale (EDSS) score of 0.0–5.0 and had experienced at least one relapse during the 12 months prior to randomization or had a Gd+ lesion within six weeks prior to randomization. The main exclusion criteria were primary or secondary progressive or progressive relapsing MS, 17 diagnosis or history of neuromyelitis optica. Only treatment-naïve Japanese patients with RRMS were included in this interim subgroup analysis.
Magnetic resonance imaging (MRI) measurements of the brain (with and without Gd+) were conducted at baseline and every four weeks during the treatment period. Magnetic resonance (MR) images were not performed within 28 days after a course of steroids to minimize any confounding effects of corticosteroids on Gd+ lesions. Validation of MRI capabilities and personnel were carried out by a central MRI reading center (NeuroRx Research) to ensure quality and consistency. MRI technicians at study sites and the central MRI reading center were blinded to patients’ treatment assignments.
The study was conducted in accordance with the Declaration of Helsinki and complied with guidelines and regulations (local and international). The study protocol was approved by the ethics committees of the participating institutions. All participants provided written consent before any evaluations or procedures were carried out.
Outcomes
This analysis was undertaken using the same endpoints as the APEX study, as such, the primary endpoint of Part I of this analysis was the total number of new Gd+ lesions on MRI scans at Weeks 12, 16, 20, and 24, compared with placebo. Secondary endpoints of this analysis were the cumulative number of new Gd+ lesions from baseline to Week 24 and the number of new or newly enlarging T2 hyperintense lesions at Week 24. Additional exploratory endpoints included the total number of new T1 hypointense lesions at Week 24, as well as clinical endpoints, such as EDSS and annualized relapse rate (ARR), which was defined as the total number of relapses in each treatment group divided by the total number of days on treatment for the group, and the ratio multiplied by 365. In Part II, patients undertook MRI scans at Week 48 and at each yearly visit. The total number of new Gd+ lesions, number of new or newly enlarging T2 hyperintense lesions and the number of new T1 hypointense lesions were evaluated. EDSS was evaluated at Weeks 36, 48, and every 24 weeks thereafter. The cut-off for this interim analysis was at Week 72.
Assessments of safety included reported AEs, physical and neurological examination, and laboratory tests. These assessments were carried out every four weeks in Part I of the study, except physical and neurological examinations, which were conducted every 12 weeks. In Part II, neurological examinations were carried out every four weeks until 48 weeks; physical examination and hematology were conducted every 12 weeks. After that, all assessments were carried out every 12 weeks until Week 72. Safety endpoints included incidences of AEs, serious adverse events (SAEs) and changes in clinical laboratory parameters. Lymphocyte counts were defined by the Common Terminology Criteria for Adverse Events Code (CTCAE) version 4.0 as Grade 0 (≥910/mm3), Grade 1 (≥800 to <910/mm3), Grade 2 (≥500 to <800/mm3), Grade 3 (≥200 to <500/mm3) and Grade 4 (<200/mm3). The upper limit of normal (ULN) of aspartate aminotransferase (AST) was defined as 34 and 36 U/L for females and males, respectively, the ULN of alanine aminotransferase (ALT) was 34 and 43 U/L for females and males; severe elevation of AST and ALT was defined as >10×ULN.
Statistical analysis
The total (cumulative) number of new Gd+ lesions at Week 12, 16, 20, and 24, the number of Gd+ lesions at Week 24, new or newly enlarging T2 hyperintense and T1 hypointense lesions were summarized using descriptive statistics (mean, standard deviation (SD), median, and range) for each treatment group. The total number of Gd+ lesions at Weeks 12 to 24 was analyzed by negative binominal regression, which was also used to analyze ARR over 24 weeks. The proportion of patients who relapsed was estimated as the cumulative probability of relapses from the Kaplan-Meier curve of the time to the first relapse during the study. Hazard ratios (HRs) were based on Cox proportional hazards model, adjusted for baseline EDSS (≤2.0 vs >2.0), baseline age (<40 vs ≥40) and number of relapses in the one year prior to study entry.
Results
From the APEX study population, this post-hoc analysis was conducted on treatment-naïve Japanese patients with RRMS treated with placebo (n=27) and DMF (n=25). Baseline characteristics of the study population were relatively well matched between treatment groups; however, patients in the DMF group tended to have less Gd+ lesions and lower T2 lesion volume and a longer disease duration at baseline (Table 1). During Part I, five patients in the placebo group and two patients from the DMF group discontinued treatment prematurely due to AEs and consent withdrawal; all remaining patients entered Part II (placebo/DMF: n=22; DMF/DMF: n=23).
Baseline demographic characteristics of treatment-naïve Japanese patients enrolled in APEX.

BMI: body mass index; DMF: dimethyl fumarate; EDSS: expanded disability status scale; Gd+: gadolinium-enhancing; MS: multiple sclerosis; SD: standard deviation.
Values are reported as mean±standard deviation unless otherwise stated.
an=18; bn=19; cn=26.
Efficacy
In Part I, DMF reduced the mean total number of new Gd+ lesions in Weeks 12–24 by 94% (95% confidence interval (CI): 78.8 to 98.5), compared with placebo (0.16 vs 2.82 total new Gd+ lesions, respectively; Table 2, Figure 1(a)). The onset of effect on new Gd+ lesions was apparent as early as four weeks after initiating treatment (0.20 vs 0.81; p=0.0148; Figure 2). DMF also reduced the mean total number of new/newly enlarging T2 hyperintense lesions at Week 24 by 70% (95% CI: 42.1 to 84.1; 0.84 vs 2.76, respectively; Table 2). The clinical endpoints were also improved with the risk of relapse up to Week 24 reduced by 72% (95% CI: 20.7 to 90.2; 6 vs 15 patients relapsed; Table 2) and an overall reduction in adjusted ARR of 62% (95% CI:–1.0 to 85.8; 0.55 vs 1.44; Table 2, Figure 1(b)), compared with placebo.
Summary of MRI and clinical endpoints in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis (RRMS) over 24 weeks.

APR: adjusted percentage reduction; ARR: annualized relapse rate; CI: confidence interval; DMF: dimethyl fumarate; Gd+: gadolinium-enhancing.
aOne patient experienced relapse twice.

(a) The total number of new gadolinium-enhancing (Gd+) lesions in treatment-naïve Japanese patients with RRMS treated with placebo and dimethyl fumarate 240 mg twice daily at Weeks 12–24. (b) Adjusted ARR for each treatment group. Mean±95% CI.ARR, annualized relapse rate; CI, confidence interval; DMF, dimethyl fumarate.

Total number of new gadolinium-enhancing (Gd+) lesions by study visit in treatment-naïve Japanese patients with RRMS treated with placebo and dimethyl fumarate (DMF) 240 mg twice daily.CI: confidence interval.
In Part I/II (72 weeks), unadjusted ARR reduced probability of relapse in the both group(DMF/DMF and placebo/DMF) (Figure 3(a)). The number of Gd+ lesions was also decreased in the placebo/DMF treatment group and maintained in the DMF/DMF treatment group (Figure 3(b)).

(a) Unadjusted annualized relapse rate (ARR) for Parts I (0–24 weeks) and II (24–72 weeks) of APEX, and (b) the mean total number of Gd+ lesions at Week 24 and 48 for placebo/dimethyl fumarate (DMF) and DMF/DMF treatment groups.
The risk of relapse was reduced by 72% with DMF compared with placebo (HR 0.28; 95% CI: 0.10 to 0.79) in Part I (24 weeks). As shown in the Kaplan-Meier curve, the probability of relapse was lower with DMF from as early as Week 8 (Figure 4(a)). After Week 24 (Part II), the probability of relapse increased slightly in both groups and the estimated proportion of patients with relapses at Week 72 was 0.605 (placebo/DMF) and 0.347 (DMF/DMF). Figure 4(b) shows the mean of EDSS score during the course of the study. Statistical analysis was not performed.

(a) Kaplan-Meier estimates of the proportion of patients who relapsed within 72 weeks, and (b) Expanded Disability Status Scale (EDSS) of patients in placebo/dimethyl fumarate (DMF) and DMF/DMF treatment groups over 72 weeks (mean±standard deviation (SD)).
Safety
The incidence of AEs was 86% (19/22) in the placebo/DMF group and 91% (21/23) in the DMF/DMF group while the incidence of SAEs was 18% (4/22) and 9% (2/23), respectively (Table 3). The rate of discontinuation of study drug due to an AE was 9% (2/22) in the placebo/DMF group and 4% (1/23) in the DMF/DMF group. Reasons for treatment discontinuation included abdominal pain and increased ALT/AST for patients in the placebo/DMF group and MS relapse for one patient in the DMF/DMF group. Common AEs included nasopharyngitis (59% (13/22) in the placebo/DMF group and 57% (13/23) in DMF/DMF group) and MS relapse (18% (4/22) and 30% (7/23), respectively). In the placebo/DMF group, MS relapse, abdominal pain upper and pruritus (18% (4/22) each), and abdominal pain and vomiting (14% (3/22) each) occurred. Rash (22% (5/23)) and pruritus (13% (3/23)) occurred most frequently in the DMF/DMF group.
Summary of adverse and serious adverse events in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis (RRMS) over 72 weeks.

AE: adverse event; DMF: dimethyl fumarate; MS: multiple sclerosis; SAE: serious adverse event.
Values are reported as number of patients (percentage of patients).
Clinical laboratory testing found 18% (4/22) of patients in the placebo/DMF group and 26% (6/23) of patients in the DMF/DMF group experienced Grade 1 or Grade 2 lymphocyte count reduction; none experienced a severe reduction in lymphocyte count (Table 4). In Part I of the study, two patients in the DMF group had AST and ALT elevation >10×ULN (one patient each) and one patient in the placebo/DMF in Part II had an ALT elevation >10×ULN (Table 4).
Clinical laboratory test of lymphocyte count, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in treatment-naïve Japanese patients with relapsing–remitting multiple sclerosis (RRMS) over 72 weeks.
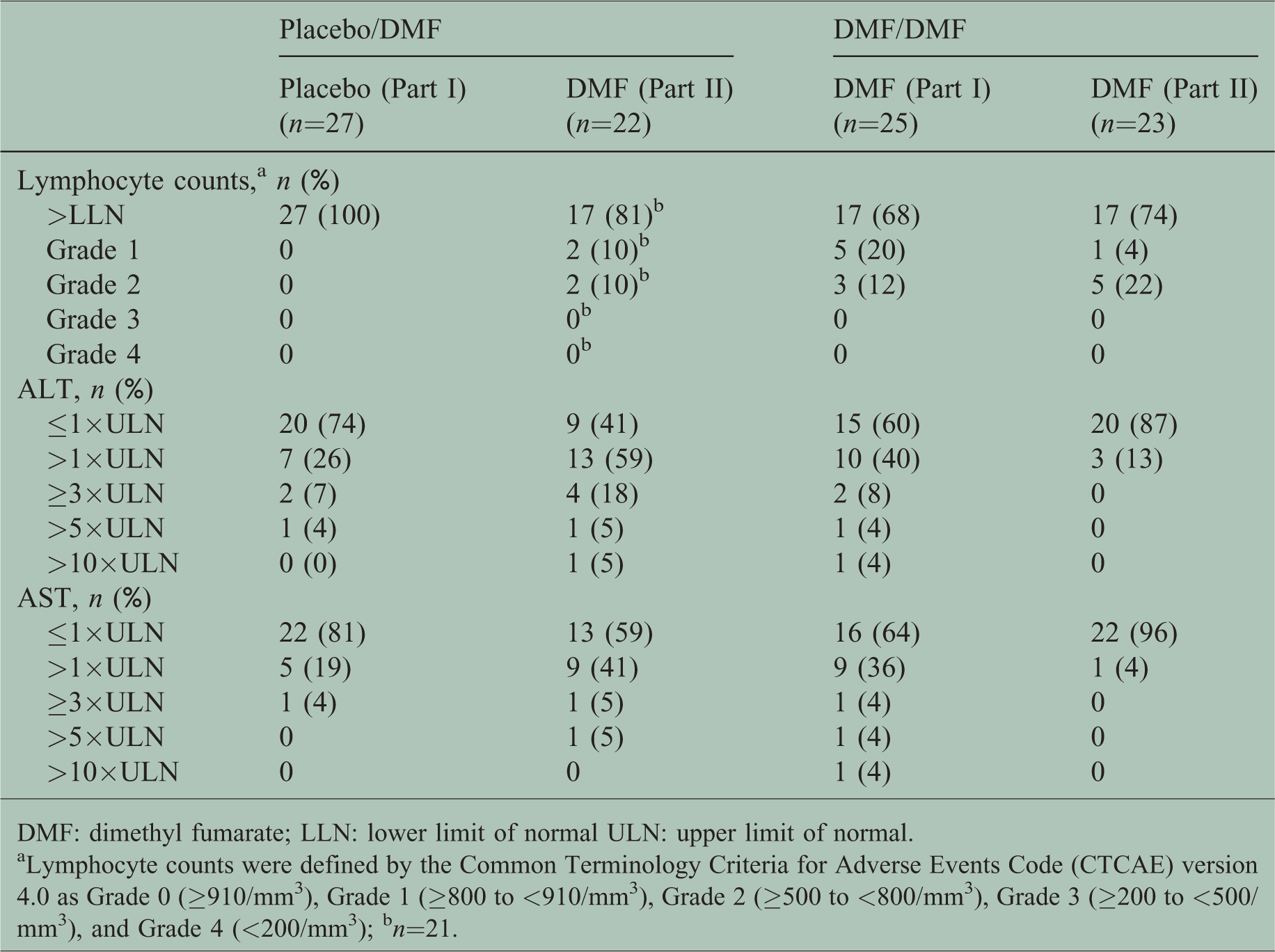
DMF: dimethyl fumarate; LLN: lower limit of normal ULN: upper limit of normal.
aLymphocyte counts were defined by the Common Terminology Criteria for Adverse Events Code (CTCAE) version 4.0 as Grade 0 (≥910/mm3), Grade 1 (≥800 to <910/mm3), Grade 2 (≥500 to <800/mm3), Grade 3 (≥200 to <500/mm3), and Grade 4 (<200/mm3); bn=21.
Discussion
This interim analysis of APEX evaluated the efficacy and safety of DMF in treatment-naïve Japanese patients with RRMS. The results demonstrated sustained efficacy of DMF on MRI lesions (Gd+ and T2) and clinical endpoints, assessed by the number of relapses throughout 72 weeks. Furthermore, the study indicated that DMF was well tolerated in this patient population.
In Part I of the study, DMF reduced total numbers of new Gd+ lesions at Weeks 12–24 (94%) and new or newly enlarging T2 hyperintense lesions at Week 24 (70%). The onset of effect on new Gd+ lesions was apparent as early as four weeks. Adjusted ARR and the risk of relapse were also reduced, by 62% (95% CI: –1.0 to 85.8) and 72% versus placebo at Week 24, respectively. The efficacy of DMF in treatment-naïve Japanese patients with RRMS observed in this subgroup analysis seemed to be comparable with prior pivotal studies. In a global phase 2b trial, patients receiving DMF 240 mg three times daily had a 69% reduction of Gd+ lesions, 48% reduction of T2 hyperintense lesions and 32% reduction in ARR at Week 24 compared with those receiving placebo. 18 Like our analysis, a subgroup analysis of the phase 2b trial showed that the Gd+ response was apparent early after initiating treatment, with a significant response observe at 12 weeks (p = 0.007). 19 Similarly, a subgroup analysis of newly diagnosed patients in the integrated data from DEFINE and CONFIRM studies reduced Gd+ and T2 hyperintense lesions by 92% and 80%, respectively. ARR and risk of relapse were also reduced by 56% and 54%, respectively. 20
The effect of DMF in this subgroup analysis of treatment-naïve Japanese population was also numerically larger than those seen in the overall Japanese population. The reduction of the total number of new Gd+ lesions at Weeks 12–24 was 85% (95% CI: 69.5 to 92.9) and the reduction in new or newly enlarging T2 hyperintense lesions was 63% in the total Japanese population. There was fewer treatment-naïve Japanese patients who experienced a relapse compared with the total Japanese population where the proportion of patients was 44% (95% CI: 13.1 to 77.4) and the ARR was 48% (95% CI: 7.4 to 71.2).
The strong efficacy of DMF in treatment-naïve patients may be explained from both a neuropathological and clinical perspective. In the early stages of disease, axonal transection, 21 increased frequency of relapse, and a higher lesion load were observed and associated with poorer long-term outcomes. 22 Hence, newly diagnosed patients may benefit from early intervention to slow the accumulation of damage and progression of disability. Furthermore, the relationship between MS disease activity and long-term clinical prognosis weakens with time, suggesting early intervention to maximize therapeutic opportunity.22,23
In this subgroup analysis, the overall incidence of AEs was similar between placebo/DMF (86%) and DMF/DMF (91%) groups. Common AEs in the DMF and placebo groups included gastrointestinal disorders, rash, and pruritus. Nasopharyngitis was also a common AE but affected placebo/DMF and DMF/DMF groups equally (59% and 57%, respectively), which suggests that the incidence may be independent of study treatment. The safety results of this subgroup analysis were similar to pivotal phase 3 studies (DEFINE and CONFIRM). The overall incidence of AEs were 96% and 94% in the DEFINE and CONFIRM trials.11,12 Common AEs found in DEFINE and CONFIRM were gastrointestinal (GI) events (31% of patients receiving placebo and 40% in patients receiving DMF), flushing and related symptoms (8% versus 45%) in the integrated analysis of these studies, and nasopharyngitis (16% versus 17%) in CONFIRM.11,24 Incidence of GI events and flushing and related symptoms were highest in the first month after DMF was initiated. 23
In this interim post-hoc part of the APEX study, AEs leading to discontinuation were limited (12% of patients total), and included one occurrence of abdominal pain and increased ALT/AST levels (placebo/DMF) and another of MS relapse (DMF/DMF). However, in the clinical practice of DMF, management of GI events and flushing is crucial for the continuation of DMF. 25 Some reports recommended patient education, dosing with food, slow titration, dose reduction, and use of symptomatic therapy to mitigate GI events.24–27
In this subgroup analysis, the proportion of patients who, post-baseline, had at least one Grade 1 or Grade 2 lymphocyte count was 26% in the DMF/DMF group and 18% in the placebo/DMF group. No Grade 3/4 lymphocyte count was observed . The majority of the patients remained within normal limits. This was consistent with currently published literature which suggested that mean and median absolute lymphocyte counts decreased by 30% during the first year of treatment and remained within normal limits. 28 The analysis suggested that being treatment-naïve may not put patients with RRMS at higher risk of a reduced lymphocyte count. In addition, the AST and ALT levels increased in the DMF group in Part I of the study and in the placebo/DMF group in Part II of the study. Three patients had AST or ALT levels exceeding 10 times the ULN in Part I or Part II of the study; however, none of these cases met the criteria for Hy’s law diagnosed by elevations in ALT or AST ≥3×ULN that were concurrent with an elevated total bilirubin >2×ULN. To optimize DMF treatment, therapeutic monitoring of adverse drug events and laboratory parameters, such as lymphocyte count and ALT/AST is advised.
The main limitation of this analysis was the limited sample size, which resulted in the lack of statistical power of clinical endpoints and reduced ability to detect rare AEs. Another limitation was the post-hoc analysis. Although clinical disease activity appeared to be well-matched between treatment arms, it seemed that there were differences between the patient groups in terms of the baseline radiological profiles. MS is a rare disease in Japan, and these limitations are expected to be addressed in the ongoing real-world studies in a wider population of Japanese patients with MS.
In conclusion, the results of this interim post-hoc analysis of treatment-naïve Japanese patients with RRMS in APEX demonstrated sustained efficacy for MRI lesions and risk of relapse throughout 72 weeks. Furthermore, the safety profile was favorable and, as such, it is appropriate to use DMF as first-line for treatment-naïve Japanese patients with RRMS.
Supplemental Material
Supplemental material for Efficacy and safety of dimethyl fumarate in treatment-naïve Japanese patients with multiple sclerosis: Interim analysis of the randomized placebo-controlled study
Supplemental Material for Efficacy and safety of dimethyl fumarate in treatment-naïve Japanese patients with multiple sclerosis: Interim analysis of the randomized placebo-controlled study by Masahiro Mori Department of Neurology, Chiba University, Japan Takashi Ohashi Department of Neurology, Tokyo Women’s Medical University Yachiyo Medical Center, Japan Yasuhiro Onizuka, Katsutoshi Hiramatsu, Hase Masakazu Biogen Japan Ltd, Japan Jang Yun, André Matta Biogen Inc., USA Shinichi Torii Biogen Japan Ltd, Japan in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Acknowledgements
Biogen Japan Ltd provided funding for medical writing support in the development of this article; Mimi Chan from inScience Communications, Springer Healthcare, wrote the first draft of this manuscript based on input from authors. Biogen reviewed and provided feedback on the paper to the authors. The authors had full editorial control of the paper, and provided their final approval of all content.
Conflict of Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MM has received speaking fees from Biogen Japan Ltd, Mitsubishi Tanabe Pharma Co. Ltd, and Takeda Pharmaceutical Co. Ltd. TO has received speaking fees and consulting fees from Biogen Japan Ltd and Takeda Pharmaceutical Co. Ltd. YO and ST are employees of Biogen Japan Ltd and stockholders of Biogen Inc. MH and KH were employees of Biogen Japan Ltd and stockholders of Biogen Inc. at the end of this study. JY was an employee of Biogen Inc. and a stockholder of Biogen Inc. at the end of this study. AM was an employee of Biogen Inc. and a stockholder of Biogen Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Biogen.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.