
Abstract
Acute kidney injury is a clinical syndrome that complicates the course and worsens clinical outcomes in patients with chronic liver diseases. It is a common complication in hospitalised patients with liver cirrhosis, especially those with decompensated cirrhosis, associated with a high mortality rate. Considering its impact on patient prognosis, efforts should be made to diagnose and tailor therapeutic interventions for acute kidney injury at an early stage. In the past decade, significant progress has been made to understand the key events and define major prognostic factors for the onset and progression of acute kidney injury in the cirrhotic population leading hepatologists to re-define the classic definition of hepatorenal syndrome and renal failure in this specific population.
Keywords
This review provides a summary of the most recent updates in the definition and management of acute kidney injury (AKI) in patients with cirrhosis.
Clinical vignette
A 42-year-old woman with a history of decompensated alcohol-related cirrhosis was admitted to the hospital emergency department for an acute episode of decompensation marked by the development of a new-onset encephalopathy and worsening ascites. At admission, a serum creatinine level of 1.4 mg/dL (124 μmol/l) was two times higher than the value obtained 3 weeks earlier. At home, she was receiving furosemide, spironolactone and propranolol and has not recently used nephrotoxic drugs. On presentation mean arterial pressure was 70 mmHg. Urine analysis showed no significant proteinuria, no haematuria, and renal ultrasonography was normal. Diagnostic paracentesis revealed a neutrophil count of 1531/μl consistent with a diagnosis of spontaneous bacterial peritonitis.
Several key clinical questions are highlighted by this case:
Regarding the increase in serum creatinine over the past 3 weeks, can we consider this clinical situation as an AKI? Are there specificities in the definition of AKI in the cirrhotic population? Is serum creatinine a good clinical marker to evaluate kidney function in this case? What are the causes of AKI in cirrhosis? How do we identify them? What are the current management strategies for cirrhotic patients with AKI? Are there any interventions to prevent AKI?
AKI defines the syndrome of acute impairment in kidney function encompassing the whole spectrum of the syndrome from minor acute reduction in markers of renal function to the requirement for renal replacement therapy. 1 For more than two centuries, there was no consensus on the diagnostic criteria or clinical definition of AKI in the general population. 1 Thus, the new concept of AKI, as defined successively by risk, injury, failure, loss of kidney function, and end-stage kidney disease (RIFLE), the Acute Kidney Injury Network (AKIN) and kidney disease improving global outcomes (KDIGO) classifications creates a new paradigm. Only a small increase (≥0.3 mg/dL (≥26.5 μmol/l)) in serum creatinine (creatinine) or a decrease in urinary output leads to increases in hospital mortality and clinical outcomes. 2
In cirrhosis, the criteria of acute renal failure in patients with decompensated cirrhosis were proposed in 1996 and were later modified in 2007. 3 , 4 Historically, the abrupt increase in creatinine of 50% or greater from baseline to a final value of 1.5 mg/dL (133 μmol/l) or greater was the most commonly accepted criterion to define acute renal failure in patients with cirrhosis. Since then, several studies have shown that the cut-off value of creatinine of 1.5 mg/dL (133 μmol/l) has a strong prognostic impact, both in terms of the resolution of AKI and survival. 5 , 6 However, this fixed cut-off encounters two main drawbacks. 7 First, it does not take into account dynamic changes in creatinine that occur during the previous days or weeks, which are critical to differentiate acute from chronic kidney disease. Second, the use of a creatinine level of 1.5 mg/dL (133 μmol/l), which corresponds to a very low glomerular filtration rate (GFR) of approximately 30 ml/min, excludes less severe AKI from the definition. 5 , 7
Therefore, in 2015, the International Club of Ascites (ICA) has adopted a new classification of AKI, derived from the KDIGO definition proposed in the general population (Table 1). 1 , 7 ICA defines AKI (grade 1) in cirrhosis as an abrupt (within 48 hours) reduction in kidney function defined as an absolute increase in creatinine of 0.3 mg/dl or greater (≥26.5 μmol/l) or a percentage increase in creatinine of 1.5–2 times from baseline in less than 7 days. Grade 2 AKI is defined by an increase in creatinine to more than 2 to 3-fold from baseline (in <7 days) and grade 3 by an increase to more than 3-fold from baseline or creatinine of 4 mg/dL or greater (≥354 μmol/l) with an abrupt increase of at least 0.3 mg/dL (≥26.5 μmol/l) or the need for renal replacement therapy. In contrast to KDIGO guidelines, the use of urinary output is not considered in the cirrhotic population as a criterion of AKI. Indeed, many factors can affect urinary output interpretation in the cirrhotic population. Urinary output can be preserved in the presence of renal dysfunction, due to maintained diuretics or, by contrast, despite preserved GFR, urinary output can be low due to avid sodium retention secondary to neurohumoral activation of endogenous vasoconstrictors in patients with refractory ascites. 8 Finally, urine collection may be challenging in this population. Even if a recent study has shown that not considering urinary output may result in an underestimation (58% vs. 82%) of AKI incidence in critically ill patients with cirrhosis, urinary output has been poorly investigated in cirrhosis. 8 , 9
Diagnostic criteria and definitions of AKI in patients with cirrhosis.
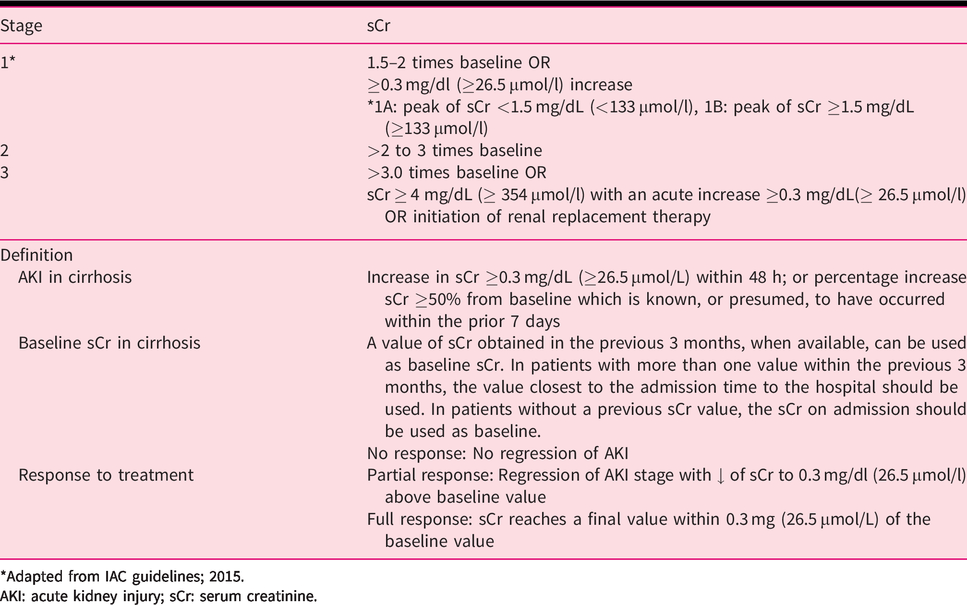
*Adapted from IAC guidelines; 2015.
AKI: acute kidney injury; sCr: serum creatinine.
The definition of baseline creatinine remains a critical issue in clinical practice. Considering the ICA definition, a creatinine baseline obtained less than 7 days prior to the admission is the ideal scenario. However, in ‘real life’, creatinine is rarely known as close as one week (see clinical vignette). Regarding the ICA guidelines and considering evidence, it is reasonable to consider the lowest creatinine within 3 months before admission as the creatinine baseline.
A recent large retrospective study showed that the closest pre-admission creatinine and the lowest creatinine within 3 months before admission shared a similar hazard ratio for mortality. 10 However, in the absence of a previous creatinine value, the creatinine at admission is considered as baseline.
The GFR is the widely accepted index of kidney function in cirrhotic and non-cirrhotic populations. 1 Creatinine remains the most commonly used clinical marker to estimate (or calculate) GFR. However, all the creatinine-based estimated GFRs used in patients with liver disease overestimate the gold standard measured GFR.11–13 Indeed, patients with decompensated cirrhosis tend to have a low creatinine level due to several factors, including low muscle mass, protein malnutrition, low creatinine production by the liver but also increased tubular secretion of creatinine and the increased volume of distribution in the setting of fluid overload. 13 , 14 In addition, in patients with high serum bilirubin levels, Jaffe’s method used for creatinine measurement could be biased due to interference with bilirubin, and enzymatic methods (e.g. bilirubin oxidase) should be preferred. 15 The modified diet in renal disease 6 (MDRD6) equation seems to be the most accurate and validated estimate to predict GFR. 16 The Royal Free Hospital cirrhosis GFR equation (using creatinine, blood urea nitrogen, international normalised ratio, gender, serum sodium, the presence of moderate/severe ascites) outperforms other equations for GFR prediction yet should be validated in larger populations. 17
Once the diagnosis of AKI has been made, identification of the cause of AKI represents a critical issue (Table 2). The most common cause of AKI in cirrhosis is pre-renal AKI, accounting for approximately 50–66% of cases. 18 , 19 Pre-renal causes are classified according to the response to volume expansion (Figure 1). In patients who are volume responsive, AKI usually results in hypovolemia due to gastrointestinal haemorrhage, aggressive diuresis, diarrhoea, or large volume paracentesis. By contrast, the absence of response to volume expansion defines the hepatorenal syndrome type of AKI (HRS-AKI), formerly called ‘type 1 hepatorenal syndrome’. Interestingly, the diagnostic criteria of hepatorenal syndrome have been revised by the ICA in 2015 (Figure 2) and the new name of type 1 hepatorenal syndrome is now HRS-AKI. 20
Causes of AKI in cirrhosis.

*The purely functional nature of HRS and the absence of renal parenchymal damage has never been definitively proved. Recent studies based on biomarkers suggest than HRS can be associated with some degree of parenchymal damage.
**ACS as defined by increased intra-abdominal pressure to greater than 20 mmHg secondary to tense ascites, may lead to AKI by increasing venous pressure resulting in compromise of microvascular blood flow. Therapeutic paracentesis in combination with albumin infusion has shown improvement in renal function in cirrhosis patients with ACS.
***Bile cast nephropathy can cause ATN in patients with liver failure in the setting of hyperbilirubinemia (mostly > 25 mg/dL) by epithelial injury in distal nephron segment, and by obstructive biliary cast formation in the tubules.
AC: alcoholic cirrhosis; ACS: abdominal compartment syndrome; AKI: acute kidney injury; ATN: acute tubular necrosis; CMV: cytomegalovirus; EBV: Epstein–Barr virus; GLS: glomerulonephritis; HBV: hepatitis B virus; HCV: hepatitis C virus; HRS: hepatorenal syndrome; PPI: proton pump inhibitors; NSAIDs: non-steroidal anti-inflammatory drugs; ++: mostly.
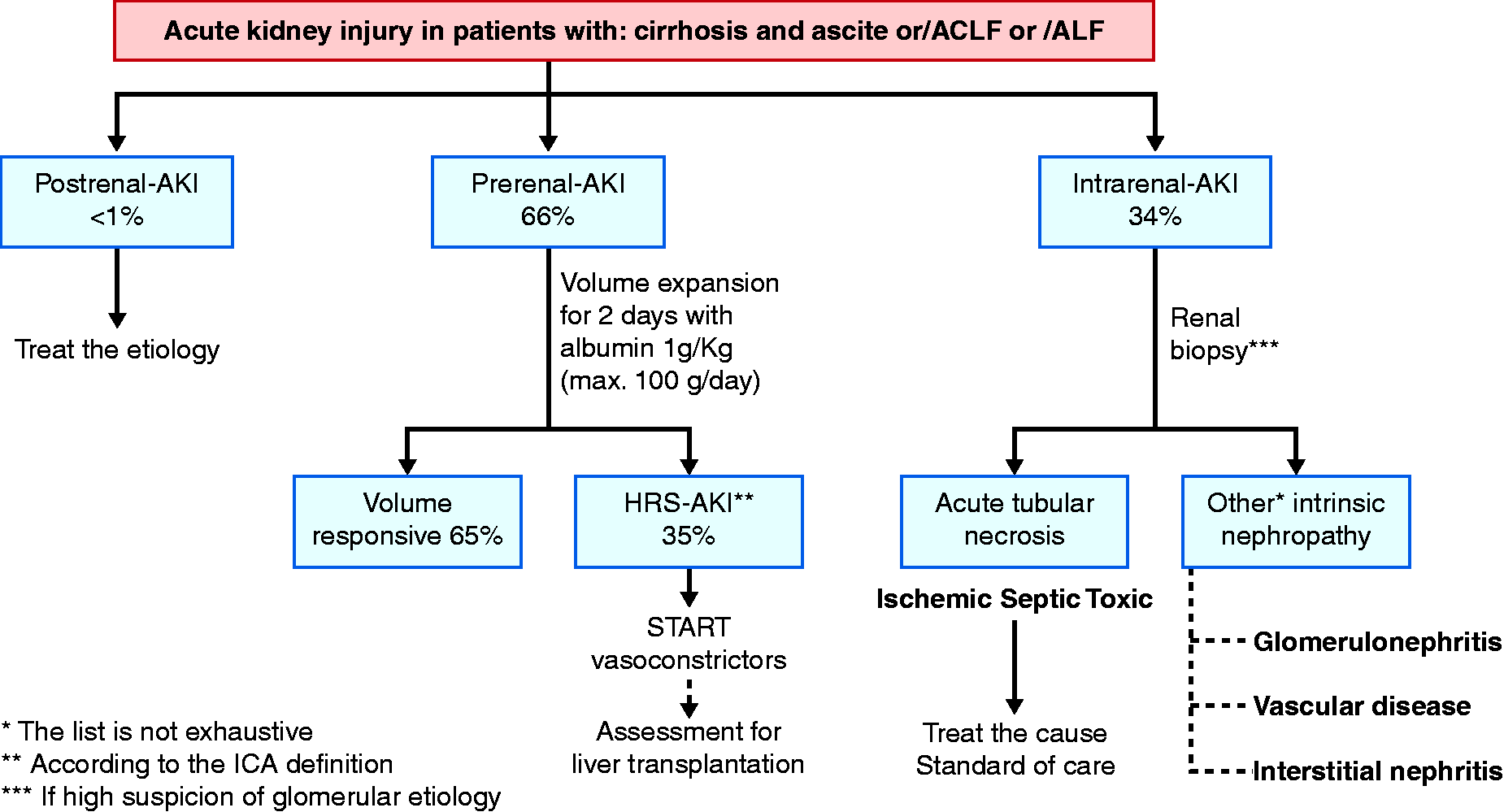
Diagnostic approach of AKI in patients with cirrhosis. ACLF: acute-on-chronic liver failure; AKI: acute kidney injury; ALF: acute liver failure.

Diagnostic criteria of HRS-AKI. NSAIDs: non-steroidal anti-inflammatory drugs; sCr, serum creatinine.
Post-renal AKI in the cirrhotic population is very rare, accounting for less than 1% in several series. 18
Acute tubular necrosis represents the second most common cause of AKI in cirrhosis ranging between 15% and 35%. 6 , 21 Acute tubular necrosis is the most common type of intrinsic AKI and is characterised by an acute impairment in kidney function caused by renal tubular dysfunction. 22 Common causes of acute tubular necrosis are precipitated by ischaemic (e.g. bleeding, diarrhoea, vomiting, renal losses via diuretics or osmotic diuresis), toxic event or sepsis. 1 Pre-renal AKI and ischaemia-induced acute tubular necrosis share the same spectrum of causes; however, in clinical practice most pre-renal AKI is resolved by plasma expansion. 23
AKI is rarely due to other intrinsic causes of the kidney, such as acute glomerulopathies (virus-associated or IgA nephropathy), vascular disease, tubular damage due to bile cast nephropathy or acute interstitial nephritis due to medications such as antibiotics/non-steroidal anti-inflammatory drugs (NSAIDs). 19 Nevertheless, these causes justify systematic urine protein dosage and sediment analysis in clinical practice. When the urine examination shows a high suspicion for glomerular diseases renal biopsy is often required for diagnosing and treating the exact aetiology.
Practically, after ruling out volume responsive pre-renal AKI by appropriate volume expansion/withholding diuretic therapy, and after having excluded specific glomeruli injury (haematuria/proteinuria), the real clinical challenge remains on the differential diagnosis between acute tubular necrosis from HRS-AKI. This major issue relies on the fact that HRS-AKI may theoretically reverse with vasopressor and albumin while no specific treatment for acute tubular necrosis exists. No conventional biomarkers available in clinical practice can accurately distinguish HRS-AKI from acute tubular necrosis in advanced cirrhosis. 23 , 24
In the setting of AKI in patients without liver disease, fractional excretion of sodium less than 1% suggests a pre-renal AKI, while a value greater than 1% is suggestive of intrinsic AKI. However, virtually all patients with advanced cirrhosis, even patients with intrinsic AKI such as acute tubular necrosis have a fractional excretion of sodium below 1%. Several studies in patients with chronic liver disease have found that the fractional excretion of sodium, with a new lower cut-off of 0.2%, may be clinically relevant to differentiate HRS-AKI (<0.2%) from acute tubular necrosis (≥0.2%) in non-volume-responsive patients. 23 Nonetheless, the clinical use of the fractional excretion of sodium in this context should be validated in future prospective studies.
Nevertheless, most recent studies describe the presence of an increased level of tubular injury biomarkers in patients with HRS-AKI, suggesting a continuum between acute tubular necrosis and HRS-AKI rather than two well-dichotomised entities. 18
General management
The first step in the management of AKI consists of identifying and promptly treating precipitating factors. 7 , 25 The most common triggers are sepsis, especially spontaneous bacterial peritonitis, but also any other infections (bacterial, fungal). 26 Less frequent precipitating factors for the development of AKI other than infection are gastrointestinal bleeding, nephrotoxic drugs, NSAIDs and iatrogenic acute kidney injury (e.g. diuretic-induced hypovolemia, excessive lactulose dose, large-volume paracentesis). 27
Acute kidney injury management discussed below is summarised in Figure 3. Irrespective of the stage, diuretics and beta-blockers should be discontinued. In the setting of pre-renal AKI, lactulose (if severe diarrhoea) should be promptly discontinued. Optimising the intravascular status is essential to prevent a worsening of renal function and to avoid complications of aggressive fluid resuscitation (e.g. worsening ascites, pleural effusion and heart failure). However, volume status assessment is particularly challenging in the context of cirrhosis. Conversely to the hypervolemic state, the patient with cirrhosis generally remains with a low effective circulatory volume (40–50% of extracellular fluid volume can be in the microcirculation). 28 To date, there is no effective monitoring tool to assess volume status in the cirrhotic population. 28 Fluid management strategies should be tailored regarding the aetiology and the severity of volume depletion. Crystalloid solutions are the first choice for patients with diarrhoea, emesis, or over-diuresis, whereas patients with acute gastrointestinal haemorrhage should receive red blood cell concentrates for a target haemoglobin level ranging between 7 g/dL or greater and less than 9 g/dL. 29 Balanced salt solutions such as Plasmalyte® may be preferred in patients with hyperchloremic acidosis. 29 A goal mean arterial pressure of 60–65 mmHg is an appropriate target in critically ill patients. 30 When volume resuscitation fails to achieve the desired mean arterial pressure, the used of vasoactive drugs is required (e.g. norepinephrine, vasopressin).
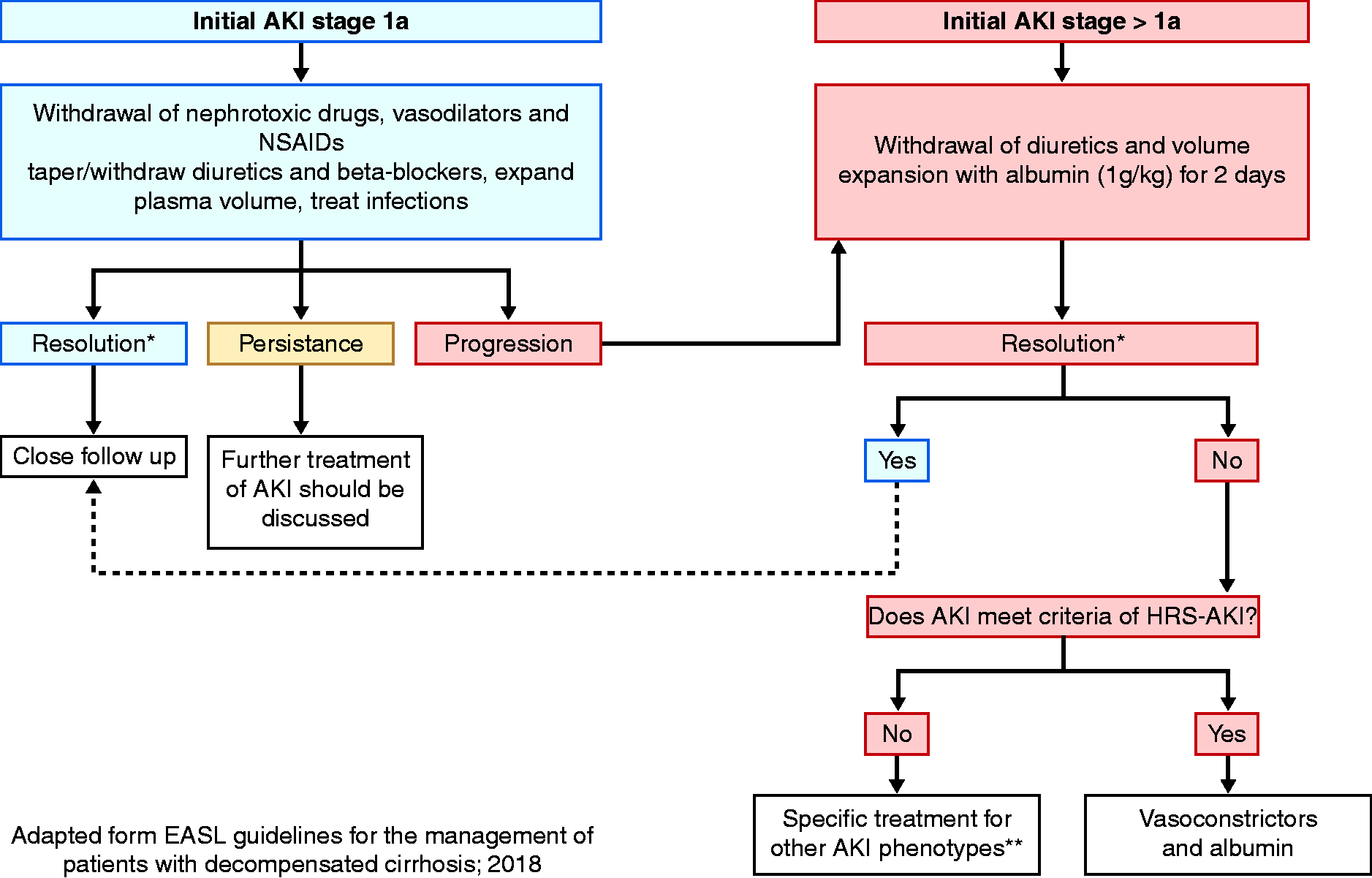
Algorithm for the management of acute kidney injury AKI in patients with cirrhosis.
All patients with AKI should be promptly evaluated for infections (e.g. chest X-ray, ascites analysis, urinalysis, etc.) and should be treated with antibiotics if there is a high suspicion of active infection. 31 Early empiric antibiotic treatment should be based on local epidemiology and resistance profiles. 32
In all cases of AKI stage over 1A, or in patients with spontaneous bacterial peritonitis-induced AKI, an intravenous albumin solution at the daily dose of 1 g of albumin/kg of body weight (not exceeding 100 g of albumin) for two consecutive days should be given. 7 In clinical practice, all cirrhosis patients with ascites diagnosed to have pre-renal AKI according to the ICA guidelines without full response (see Table 1) after 2 days of volume expansion therapy should be managed as HRS-AKI. 23
Pharmacological treatment of HRS-AKI
The cornerstone of the initial management of HRS-AKI is fluid resuscitation with albumin in combination with the early administration of vasoactive drugs. By counteracting the splanchnic arterial vasodilation associated with HRS-AKI, vasoconstrictors improve the renal circulation. 20 In the setting of HRS-AKI, terlipressin remains the most commonly used vasoconstrictor. The response rate to terlipressin plus albumin in the most recent randomised clinical trials ranges from 20% to 80% with a 3-month overall survival ranging between 27% and 58%. 8 , 33 , 38 , 39 The discrepancy in the reported response rate to terlipressin has probably been influenced by the degree of inter-trial heterogeneity. 40 Nevertheless, to our knowledge, a small number of randomised controlled trials are based on new criteria for HRS-AKI. 41 Several trials have shown a trend towards improving short-term survival in the terlipressin arm compared with placebo. 42 However, considering the poor clinical outcome in patients who developed HRS-AKI, terlipressin should be mostly considered as a bridge to liver transplantation.
Initial terlipressin therapy needs to be tailored to optimise the response rate while avoiding serious side effects. Terlipressin can initially be started as a slow intravenous bolus dose (0.5–1 mg every 4–6 hours) and should be continued for the first 3 days. After 3 days of treatment, if creatinine has decreased by 25% or more from the baseline value the treatment regimen should be maintained until a complete response (creatinine <1.5 mg/dL (<133 μmol/l)) is achieved or up to 14 days either in the case of partial or non-response. Partial response is defined by a 50% or greater improvement in creatinine from baseline without falling below 1.5 mg/dL (133 μmol/l). Whether creatinine has decreased by less than 25% on day 4, terlipressin can be progressively increased to 2 mg every 6 hours. The concomitant administration of albumin at a dose of 1 g/kg body weight on the first day followed by 20–40 g/day is recommended. Recently, a randomised controlled trial showed that the administration of terlipressin by continuous infusion (initial dose of 2 mg/day to maximum dose of 12 mg/day) has a better safety profile, with fewer severe treatment-related adverse events (20.59% vs. 43.25%, P<0.05) than its administration by bolus with a similar response rate. 43 These results are possibly explained by differences between the prolonged half-life of terlipressin (6 hours) and its short time effect on portal pressure (3–4 hours). The most common side effects of terlipressin include abdominal cramps and diarrhoea. Severe adverse events encompass cardiovascular ischaemic complications and circulatory overload. 44 The rates of treatment discontinuation due to adverse events are approximately 20%. 44
Besides terlipressin, two vasoactive drugs remain available for the treatment of HRS-AKI in clinical practice: intravenous noradrenaline or midodrine plus octreotide.
A recent meta-analysis showed that noradrenaline may have a beneficial effect regarding the HRS-AKI reversal rate without a significant improvement in short-term mortality. 40
A recent open-label randomised trial enrolling patients with acute-on-chronic liver failure diagnosed with HRS-AKI has shown that terlipressin used as an infusion was superior to noradrenaline in the management of HRS-AKI, and it should be considered as the first-line therapy in this specific population. 41
However, large randomised controlled trials are required to demonstrate the efficacy of noradrenaline for main clinical outcomes, including the reversal rate of HRS-AKI and transplant-free survival. 40
Where terlipressin is not available (mostly in the United States), noradrenaline is the best option. In contrast to terlipressin, noradrenaline infusion should be made into a central venous line under close monitoring, and therefore its use should be restricted to intensive care units. 23 In other cases, oral midodrine plus subcutaneous or intravenous octreotide represents a safe alternative. 40 Nevertheless, low-quality evidence supports the latter therapeutic option over placebo.
Non-pharmacological therapies include transjugular intrahepatic porto-systemic shunt (TIPS), renal replacement therapy and liver support systems.
It has been suggested in small uncontrolled studies or case reports that TIPS may improve renal function in patients with HRS-AKI; 45 however, TIPS is usually contraindicated due to liver insufficiency in these patients and should not be recommended. Renal replacement therapy may be proposed in non-responders to vasoconstrictors. Only a few studies have reported the results of renal replacement therapy in cirrhosis and renal replacement therapy is still debated. Indications are similar to those proposed in the general population, including severe and/or refractory electrolyte or acid-base imbalance, severe or refractory volume overload and/or symptomatic azotemia. Currently, renal replacement therapy should only be proposed in candidates for liver transplantation and is considered to be a bridge. 46 Continuous renal replacement therapy is preferred as it is probably better tolerated. The molecular adsorbent recirculating system (MARS) or Prometheus have been proposed in HRS-AKI; however, they failed to demonstrate any benefit and are not currently recommended.
Liver transplantation
Liver transplantation is the best therapeutic option in patients with HRS-AKI because it theoretically reverses renal impairment by restoring normal systemic and splanchnic circulation. However, data indicate that about 17–25% of HRS-AKI patients do not achieve HRS-AKI reversal after liver transplantation. 47 , 48 Recognised risk factors for worse outcomes after liver transplantation in patients with HRS-AKI are higher pretransplant creatinine levels, a longer duration of HRS-AKI and a longer duration of pretransplant dialysis. Wong et al. reported a 6% increased risk of non-reversal HRS-AKI with each additional day of renal replacement therapy. 48 Several studies have shown that overall death rates after liver transplantation were higher for HRS-AKI patients than non-HRS-AKI patients. 49
Simultaneous liver and kidney transplantation can be a therapeutic option for patients with cirrhosis presenting with sustained AKI. In contrast to patients listed for liver transplant with established chronic kidney disease, the criteria for simultaneous liver and kidney transplantation allocation among patients with cirrhosis and severe AKI are quite heterogenous. Currently, there are no accurate tools to predict renal recovery after liver transplantation.
The European Association for the Study of the Liver (EASL) practice guidelines recommend simultaneous liver and kidney transplantation for cirrhosis patients with severe acute renal impairment in the following conditions: (a) an estimated GFR or calculated by creatinine clearance of 25 ml/min or less for 6 weeks; or (b) AKI requiring renal replacement therapy for more than 4–6 weeks. 29
Prevention of AKI is a key issue in the management of patients with cirrhosis. It is based on the avoidance of nephrotoxic agents, the prevention of infections and of hypovolemia.
Non-steroidal anti-inflammatory agents and renin–angiotensin–aldosterone system blockers could affect intra-renal blood flow and should be avoided. Similarly, antibiotics such as vancomycin, aminoglycosides, or amphotericin B and radiocontrast agents could have direct renal tubule toxicity and should be used with caution. 29 There is no strong evidence that the cirrhotic state is a risk factor for contrast-associated AKI; however, contrast agents should be administered with caution, especially in critically ill patients or those with known chronic kidney disease. 50 Finally, drugs that could induce allergic interstitial injury should be avoided.
Antibiotic prophylaxis of spontaneous bacterial peritonitis as well as systematic antibiotics in patients with variceal bleeding are known to prevent AKI. 51 Similarly, any situation of hypovolemia promptly requires volume replacement to prevent AKI, such as blood red cells in bleeding, albumin in large volume paracentesis, or in patients with spontaneous bacterial peritonitis. Patients with cirrhosis and spontaneous bacterial peritonitis should be promptly treated by antibiotics plus albumin (1.5 g/kg body weight at diagnosis followed by 1 g/kg on day 3). 52 Regarding two recent randomised clinical trials with decompensated cirrhosis and non-spontaneous bacterial peritonitis infection, human albumin infusion failed to improve survival. 26 , 53 Therefore, routine human albumin infusion is currently not recommended in non-spontaneous bacterial peritonitis infection.
In patients with alcoholic hepatitis, N-acetyl-cysteine may prevent HRS-AKI and could be given. 54
By decreasing cardiac output, non-selective beta-blockers, which are widely used in this population, may precipitate AKI by decreasing renal blood flow. While non-selective beta-blockers are widely used to decrease portal hypertension and improve survival, they have been shown to be associated with more pronounced paracentesis-induced circulatory dysfunction and reduced survival in patients with refractory ascites. In these patients, Baveno VI recommendations propose stopping beta-blockers. 55
Conclusion
AKI is a common complication in patients with chronic liver diseases, especially those with decompensated cirrhosis, associated with a high mortality rate. In the new HRS-AKI criteria, dynamic changes in creatinine value are considered, thus helping to identify and treat HRS-AKI earlier, a key issue to improve the prognosis.
For many years, the key issue was to differentiate acute tubular necrosis from HRS-AKI because both prognosis and treatment differ. Unfortunately, no robust tool has yet emerged in the literature. In the future, the identification of markers of reversibility/irreversibility of renal dysfunction may be an interesting approach, kidney fibrosis markers being the most promising.
Footnotes
Author contributions
LOS and CF designed the review. LOS and CF drafted and wrote the review. Both authors approved the final version of the manuscript to be published.
Declaration of conflicting interests
The author(s) have no conflicts of interest to declare.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: LOS is currently a research fellow from Fund for Scientific Research (FRS-FNRS). LOS is supported by Fonds Erasme.
Informed consent
Informed consent not required as no patients were included.