
Abstract
Background
Adherence to mesalazine treatment is essential for the successful treatment of ulcerative colitis.
Objective
The objective of this study was to compare the efficacy, safety and preference of a novel high-dose 1000 mg mesalazine tablet versus conventional treatment for ulcerative colitis remission.
Methods
This pivotal phase III trial compared one 1000 mg mesalazine tablet (M1000 group) versus two registered 500 mg mesalazine tablets (M2x500 group), both taken three times daily, in patients with mild to moderately active ulcerative colitis. The primary efficacy variable was clinical remission at week 8.
Results
A total of 306 patients were considered for intent-to-treat analysis. Clinical remission was achieved in 45.0% of the patients in the M1000 group versus 41.9% in the M2x500 group (P < 0.001 for non-inferiority). Mucosal healing was achieved by 68.9% of the patients in the M1000 group and 68.4% in the M2x500 group. The majority of patients preferred the intake of one high-dose tablet (47.7%) over two low-dose tablets (10.5%). Oral treatment with high-dose 1000 mg mesalazine tablets was well tolerated without new safety signals.
Conclusions
The novel high-dose 1000 mg mesalazine tablet is effective, non-inferior to the registered 500 mg mesalazine tablet, and safe for ulcerative colitis treatment. It was preferred by a majority of patients and may improve ulcerative colitis treatment adherence.
Introduction
Ulcerative colitis (UC) is a chronic relapsing and remitting condition characterised by diffuse inflammation of the colon and rectal mucosa causing the hallmark symptom of bloody diarrhoea. Clinical presentation may vary, 1 but a range of symptoms and systemic effects significantly compromise quality of life and work productivity 2 as the severity of clinical or endoscopy disease activity increases. 3
The choice of intervention depends on the severity, localisation and pattern of disease. However, mesalazine at a dose of over 2 g/day, combined with a rectal mesalazine preparation (1 g/day in the form of a suppository, enema or foam preparation), is the recommended treatment for the induction of remission in mildly to moderately active UC. 4 A meta-analysis of randomised, double-blind studies of delayed-release preparations has confirmed a significant benefit versus placebo for the induction of clinical and endoscopic remission or improvement of disease.5,6
An important challenge to the successful treatment of UC is non-adherence to the prescribed regimen. One large-scale analysis reported adequate adherence to mesalazine therapy over a one-year period in fewer than 30% of patients, although adherence was higher (∼40%) with delayed or extended-released preparations. 7 Even during acute flares, 6–8% of patients have shown non-adherence. 5 A higher number of pills is associated with non-adherence in inflammatory bowel diseases generally 8 and UC specifically, 9 with patients expressing a preference for fewer pills.9,10
A novel high-dose 1000 mg mesalazine tablet was developed to simplify the daily drug regimen and to increase adherence to the prescribed therapy by reducing the daily pill burden and by improving the patient’s ability to swallow the tablets. Like the registered Salofalk 500 mg gastro-resistant tablets, the high-dose tablet was developed as an enteric-coated dosage form using Eudragit polymers to facilitate a pH-dependent release profile. Importantly, and despite the fact that twice the amount of drug substance is present per unit, the novel 1000 mg tablets are not double the size of the registered 500 mg mesalazine tablets.
The current study compared the efficacy, safety and patients’ preference of a single novel high-dose 1000 mg mesalazine tablet (M1000 group) versus conventional treatment with two registered 500 mg tablets (M2x500 group), both given three times daily, for remission of mild to moderately active UC. The primary objective was to prove non-inferiority of the 1000 mg tablet versus the registered 500 mg tablet in terms of achieving clinical remission.
Methods
Study design and conduct
This was a double-blind, double-dummy, randomised, multicentre, eight-week phase III study conducted during January 2013 to October 2014 at 42 gastroenterology centres in Germany, Hungary, Latvia, Lithuania, Poland, Russian Federation and Ukraine (EudraCT number 2012-001830-32; ClinicalTrials.gov identifier NCT01745770). The study was conducted in accordance with the principles of good clinical practice, the Declaration of Helsinki, and all applicable national laws, following approval by competent authorities and independent ethics committees for all participating centres. Written informed consent was obtained from all patients before the start of the study. Relevant study data were recorded and analysed pseudonymously, i.e. without patient’s name and address.
Eligibility
Patients aged 18–75 years were eligible to take part in the study if they had endoscopically and histologically confirmed active UC, with a clinical activity index (CAI) >4 and ≤12 and endoscopic index (EI) of 4 or greater, and if the extent of disease was more than 15 cm ab ano. Key exclusion criteria were Crohn’s disease, other forms of colitis, coeliac disease, malabsorption syndromes, screening stool positive for infections causing bowel disease, current relapse occurring during maintenance therapy with mesalazine more than 2.4 g/day, treatment with immunosuppressant drugs within three months and/or corticosteroids (oral, intravenous or topical rectal) within four weeks prior to baseline and abnormal renal or liver function.
Study drug and concomitant medication
Random assignment of eligible patients was performed by a computer-generated randomisation list with an allocation ratio of 1:1. Random assignment took place using randomly permuted blocks, which were used to dispense the study drug to the investigating centre. Patients were randomly assigned either to a single 1000 mg mesalazine gastro-resistant tablet with two 500 mg placebo tablets (M1000 group), or to two standard 500 mg mesalazine gastro-resistant tablets (Salofalk, Dr Falk Pharma GmbH, Freiburg, Germany) with one 1000 mg placebo tablet (M2x500 group). The taste and appearance of placebo tablets were identical to the corresponding verum medication.
Study endpoints
The primary efficacy variable was clinical remission (defined as CAI ≤4, with stool frequency and rectal bleeding subscores of 0 at week 8 (last observation carried forward; LOCF) method). The CAI was calculated according to Rachmilewitz 11 as the sum of the scores of seven variables (number of weekly stools, bloody stools, abdominal pain, general wellbeing, body temperature, extra-intestinal manifestations and erythrocyte sedimentation rate/haemoglobin). Patients completed a daily diary throughout the study, and the scores for the first four variables of the CAI were based on data collected in the patient’s diary during the seven days preceding a study visit.
Secondary efficacy variables included the rate of clinical improvement (CAI decrease ≥3 from baseline to final on-treatment visit); time to first resolution of clinical symptoms; EI; histological index (HI); course of centrally measured fecal calprotectin; and patients’ quality of life, work productivity and preference for study drug.
The EI was evaluated according to Rachmilewitz, 11 with endoscopic remission (mucosal healing) defined as EI <4 at the final visit. Histological assessment was performed using the HI according to Riley et al., 12 based on the most severely inflamed segment: histological improvement was defined as a decrease of 1 point or greater from baseline values of 2, 3 or 4 to the final visit. Patients’ quality of life was measured by the short health scale (SHS). 13 Work productivity was measured by the work productivity and activity impairment (WPAI:UC) instrument. 14
Safety variables included the incidence and type of adverse events and laboratory parameters.
Evaluation schedule
The study comprised a seven-day screening period and an eight-week double-blind treatment period followed by a two-week follow-up phase. Study visits took place at screening (week –1), baseline (week 0) and weeks 2 and 4, with a final visit at week 8 (or the withdrawal visit, if earlier). Endoscopy was performed at screening and at the final visit, and the EI and HI scores were calculated. Adherence with the study drug regimen was assessed by the investigator at each post-baseline visit by counting the unused trial medication returned by the patient and comparing it with the documentation of administered medication in the patient diary. At the final visit, patient’s preference for tablet intake was documented.
Statistical analysis
The study was performed according to an adaptive three-stage group sequential design, with possible decisions to stop the trial or continue with the pre-planned or reassessed sample size at each planned interim analysis, which was prespecified in the clinical study protocol. The first interim analysis was planned after observation of 240 evaluable intent-to-treat (ITT) patients and the second interim analysis after 320 evaluable ITT patients. Total sample size was 400 evaluable ITT patients. The sample calculation assumed a remission rate of 55% in both the investigational and control arms based on recent trial results15,16 with a non-inferiority margin of 15%, based on published data. 17 An independent data monitoring committee (IDMC) was established by the sponsor upfront to review interim efficacy and safety results. After the first interim analysis, the IDMC recommended that recruitment to the study be stopped due to statistical proof of non-inferiority, according to prespecified stopping rules. The following results show only those for the final ITT and per protocol (PP) populations.
The safety population included all randomly assigned patients who received at least one dose of study drug and provided at least one post-baseline safety evaluation. The ITT population comprised all randomly assigned patients who received at least one dose of study drug. The PP population included all ITT patients who provided at least one post-baseline efficacy assessment under study drug, received study drug for 10 days or longer and were adequately compliant (defined as taking ≥80% of study drug based on patient diaries and pill counts at each study visit).
Results
Patient population
A total of 374 patients were screened for enrolment into the study. Of these, 306 patients were randomly assigned and received at least one dose of study drug (M1000: 151; M2x500: 155) and comprised the ITT and safety populations (Figure 1). Fourteen patients terminated the study prematurely, with the frequent primary reasons being lack of patient cooperation, lack of efficacy and intolerable adverse events. In total, 278 patients met the criteria for inclusion in the final PP population (M1000: 134; M2x500: 144). The most frequent reasons for exclusion from the PP population were treatment incompliance, premature discontinuation due to reasons unrelated to study drug and a time interval of more than two days between last study drug administration and final visit.
Patient disposition. PP: per protocol.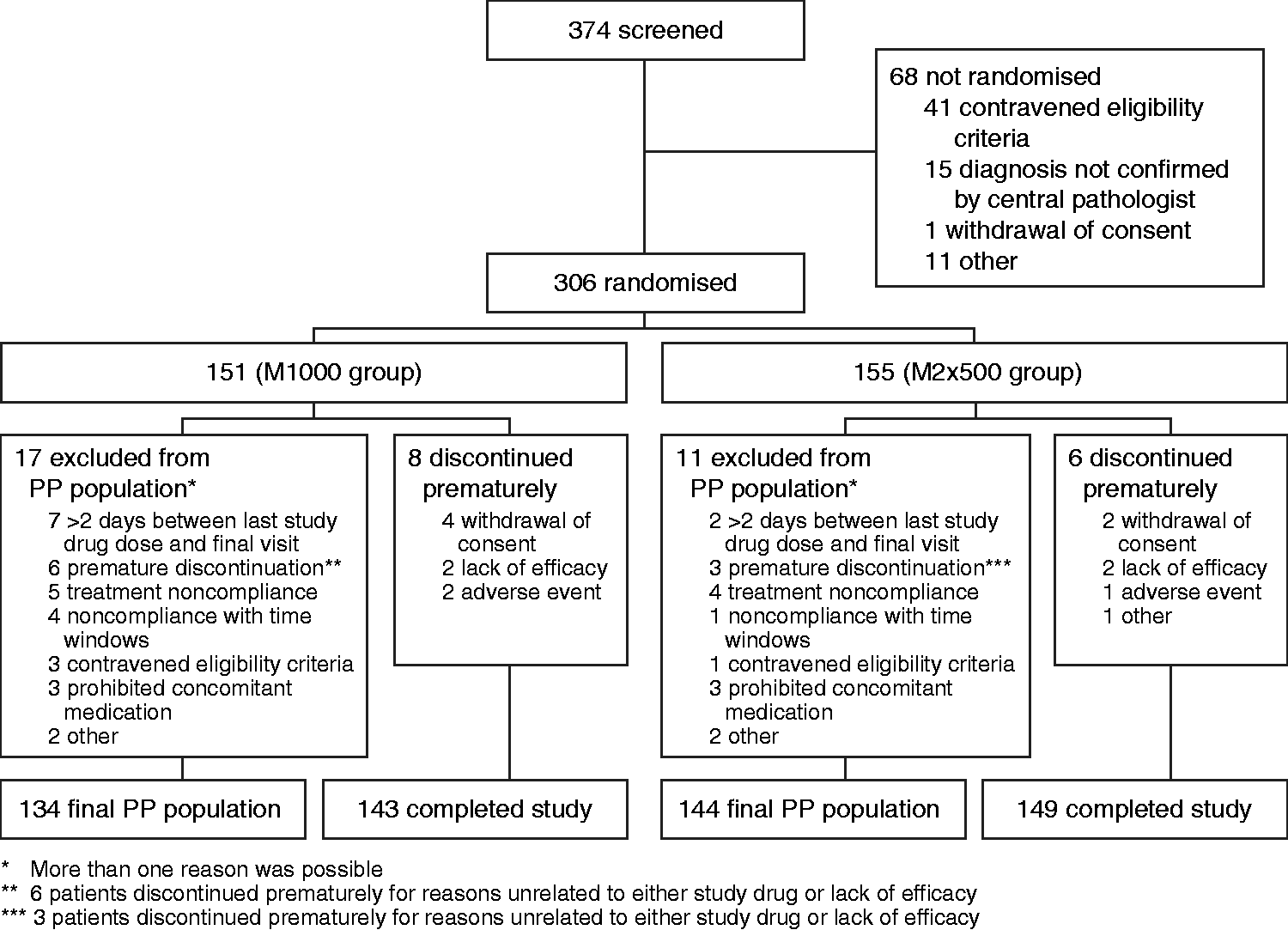
Patient demographics and baseline characteristics (final study ITT population).
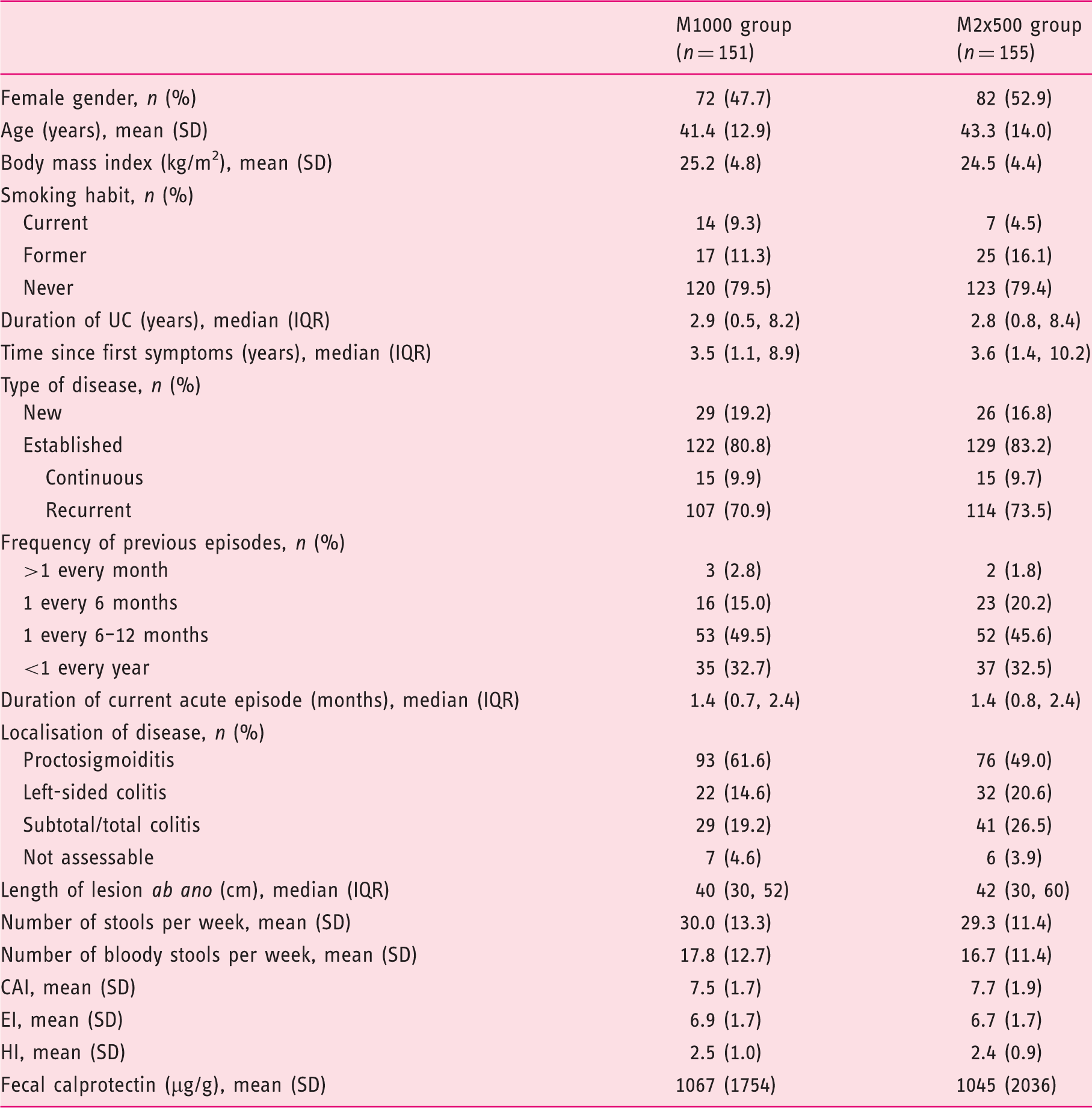
ITT: intent-to-treat; SD: standard deviation; CAI: clinical activity index; EI: endoscopic index; HI: histological index; IQR: interquartile range; UC: ulcerative colitis.
Study drug
Adherence with mesalazine and placebo (>80% of administered dose) during the eight-week study was 96.7% (146/151 patients) in the M1000 group and 97.4% (151/155) in the M2x500 group.
Primary efficacy variable
In the final ITT population, clinical remission was achieved in 45.0% (68/151) of patients receiving 1000 mg mesalazine tablets versus 41.9% (65/155) given two 500 mg mesalazine tablets (difference 3.1%; 95% repeated confidence interval (CI) −11.7%, 17.8%; P < 0.001 for non-inferiority). The analysis performed in the final PP population also confirmed this finding (Figure 2).
Clinical remission at week 8 (last observation carried forward). Results are shown for final intent-to-treat (ITT) and per protocol (PP) populations (explorative analyses).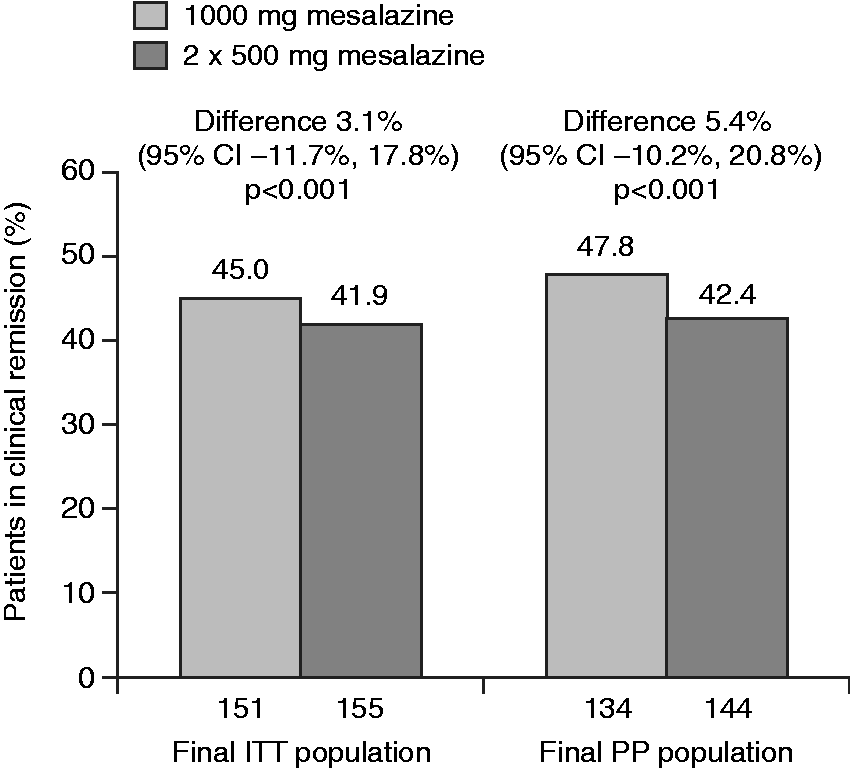
Secondary efficacy variables
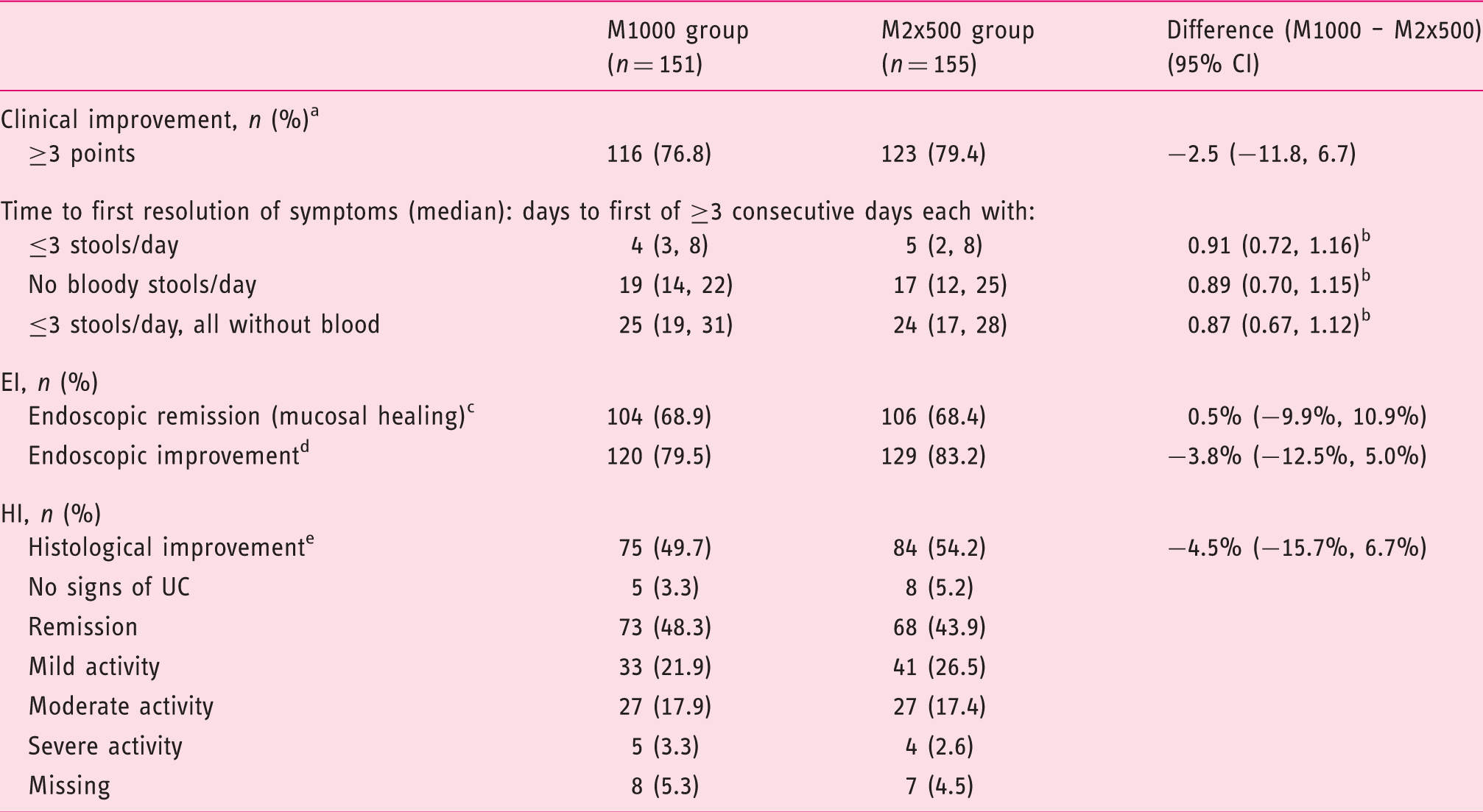
Clinical
Secondary efficacy outcomes at final visit (final study ITT population).
Decrease of ≥3 points in clinical activity index from baseline to final visit.
Hazard ratio (95% CI) for M1000 versus M2x500 group.
EI score <4.
EI score decrease of ≥1 from baseline.
HI decrease of ≥1 point from baseline values of 2, 3 or 4.
ITT: intent-to-treat; CI: confidence interval; EI: endoscopic index; HI: histological index; UC: ulcerative colitis.
The time to first resolution of symptoms according to each of the prespecified criteria was similar between both groups (Table 2).
Endoscopy and histology
Mean EI score observed at the final visit was 2.8 in the M1000 group versus 2.8 in the M2x500 group. The proportion of patients in endoscopic remission (i.e. mucosal healing, defined as EI score <4 at the final visit (LOCF)) was 68.9% in the M1000 group and 68.4% in the M2x500 group (Table 2). The difference between the two treatment groups was not statistically significant (95% CI –9.9%, 10.9%).
At the last visit, HI assessment showed that 73 patients (48.3%) in the M1000 group and 68 (43.9%) in the M2x500 group were in histological remission (Table 2). Approximately half of the patients in each group showed histological improvement according to the HI score (Table 2). The proportion of patients with no signs of UC or remission at the final visit (LOCF) was 53.0% in the M1000 group and 50.3% in the M2x500 group.
Fecal calprotectin
The mean level of fecal calprotectin, an established marker for intestinal inflammation in UC, decreased from 1067 µg/g at baseline to 746 µg/g at the final visit (LOCF) in the M1000 group, and from 1045 µg/g to 463 µg/g in the M2x500 group. The mean change from baseline was significant in both groups (M1000 −318 µg/g (95% CI −585, −50 µg/g); M2x500 −559 µg/g (95% CI −922, −195 µg/g)); the difference was not statistically significant between the two groups (241 µg/g (95% CI −210 µg/g, 692 µg/g)). The proportions of patients with fecal calprotectin levels of 50 µg/g or less or 250 µg/g or less at the final visit (LOCF) were 27.2% and 52.3% in the M1000 group, respectively, and 24.5% and 56.1% in the M2x500 group.
Patient-reported outcomes
Improvement of patients’ quality of life and work productivity at week 8 (final study ITT population)’
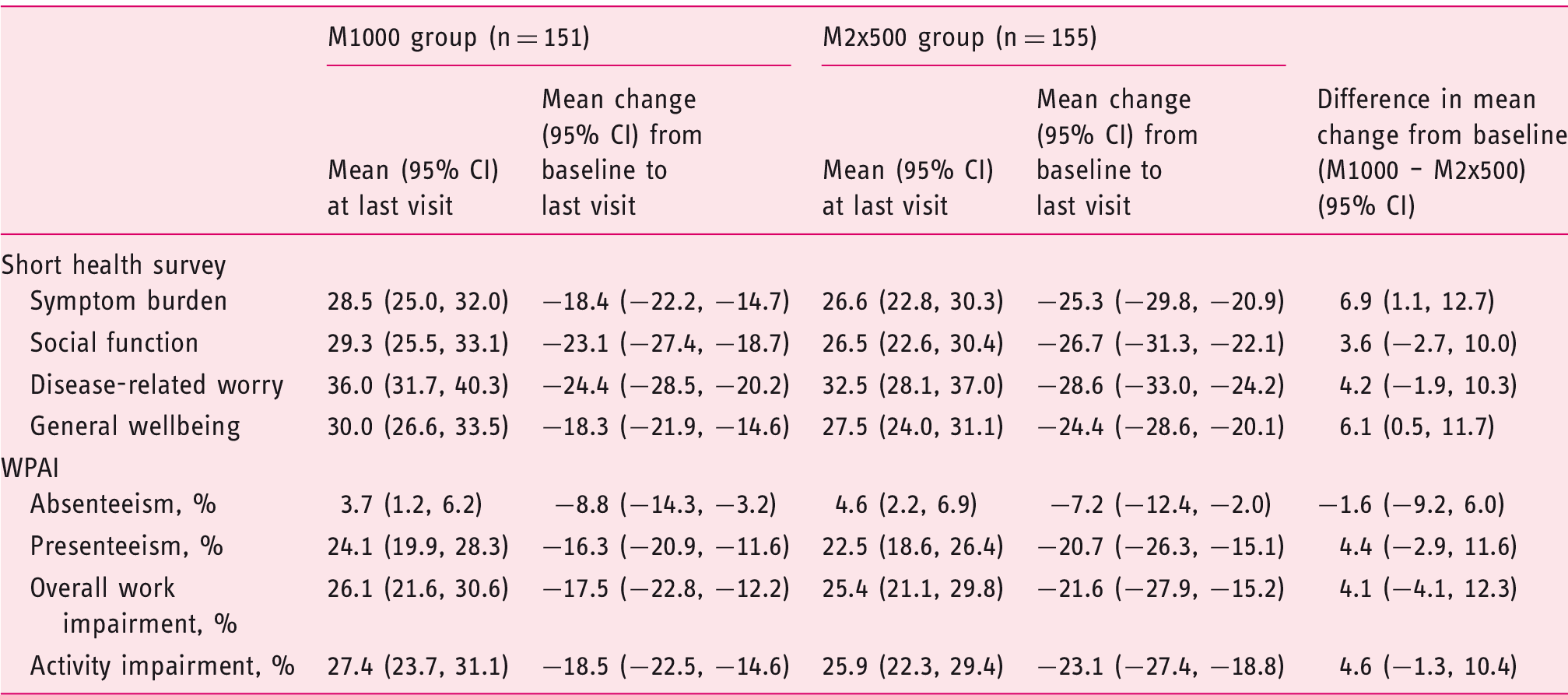
ITT: intent-to-treat; CI: confidence interval; WPAI: work productivity and activity impairment.
Absenteeism = % time missed due to ulcerative colitis.
Presenteeism = % impairment while working due to ulcerative colitis.
Work productivity, as assessed by the WPAI:UC, showed a significant improvement from baseline to final visit in both groups for each of the four scales assessed (Table 3). The change from baseline did not differ significantly between groups. Except for absenteeism, improvement in WPAI:UC correlated with an improvement in clinical activity (defined as CAI improvement ≥3 points) in the M1000 group (P < 0.001) and in the M2x500 group (P < 0.001).
Tablet preference
The proportion of patients preferring one larger mesalazine tablet was 47.7% (n = 146) compared to only 10.5% of patients who preferred two smaller 500 mg tablets (n = 32); 40.5% of patients had no preference (n = 124) (data were missing for four patients). In almost all cases, the lower number of tablets was the reason for preferring the one large mesalazine tablet (M1000 group 95.9%, M2x500 group 97.3%).
Safety
A total of 99 treatment-emergent adverse events (TEAEs) occurred in 30/151 patients (19.9%) in the M1000 group and 29/155 patients (18.7%) in the M2x500 group during the treatment phase. The most frequent of these were investigations (10 and seven patients, respectively), gastrointestinal disorders (seven and nine patients, respectively) and infections and infestations (eight and five patients, respectively). Four patients in each group experienced at least one adverse event with a suspected relation to the study drug, with nine such events in the M1000 group (increased gamma-glutamyltransferase (GGT) (two), increased aspartate aminotransferase (AST) (two), increased alanine aminotransferase (ALT), increased alkaline phosphatase, increased transaminases, increased lipase and back pain) and eight events in the M2x500 group (increased AST, increased ALT (two), increased GGT, increased alkaline phosphatase (two), increased lipase and cholestatic hepatitis). There were no serious adverse events or deaths during the study. One TEAE was graded severe, occurring in a patient in the M2x500 group (worsening of symptom of UC, not drug related). Two TEAEs (peripheral swelling and pharyngeal oedema) in one patient in the M1000 group and two TEAEs (UC and cholestatic hepatitis) in two patients in the M2x500 group led to study drug withdrawal. Of these, one TEAE was assessed as related to the study drug (cholestatic hepatitis).
Clinical laboratory parameters did not raise a safety concern. Renal function tests using the sensitive early marker cystatin C showed no impairment of renal function.
Physicians described tolerability of the study drug as ‘very good’ or ‘good’ for 90.1% and 93.5% of patients in the M1000 and M2x500 groups, respectively. From the patient evaluation, the corresponding proportions were 90.7% and 90.3%.
Discussion
This large, double-blind, placebo-controlled randomised trial demonstrates that the novel high-dose 1000 mg mesalazine tablet offers similar efficacy compared to two standard 500 mg tablets for the induction of clinical remission in mild to moderately active UC over an eight-week period using the CAI score according to Rachmilewitz 11 as the primary endpoint. The CAI score is a standardised and established instrument for evaluating the effectiveness of interventions in UC. 18 The CAI has been used in several randomised trials in this setting.15,19–22 In order to provide better comparability to previously published clinical studies of mesalazine, the CAI according to Rachmilewitz, 11 and not the Mayo Clinic score, was chosen to assess disease activity and clinical response and clinical remission rates. The CAI includes important patient-reported outcome measures such as stool frequency, abdominal pain and blood in the stool, parameters that are all increasingly requested by the US Food and Drug Administration and the European Medicines Agency as outcome measurements in clinical trials. However, it is important to consider the tools and endpoint definitions that are used to assess response and remission in various clinical trials, as they may differ significantly and could result in different response and remission rates in trials of similar drugs.
The high rates of clinical remission (42–45%) and clinical improvement (77–79%), combined with the high rate of mucosal healing (68–69%), confirmed the suitability and relevance of oral mesalazine 3 g/day as first-line treatment in this patient setting.
The primary efficacy analysis demonstrated statistical non-inferiority for the novel high-dose 1000 mg mesalazine tablet versus two standard 500 mg tablets in terms of clinical remission. A series of secondary clinical, histological and endoscopic assessments confirmed the robustness of the primary analysis. Clinical symptoms improved rapidly and profoundly, leading to fast resolution. About 80% of patients in both groups achieved endoscopic improvement and approximately 50% showed histological improvement. Mucosal healing was achieved in the majority of patients by the final study visit, confirming the premise that mesalazine is able to induce mucosal healing in patients with mild to moderate UC, and that mucosal healing can be achieved without immunosuppressants or biological agents in this patient population.
The reduced clinical activity of UC was mirrored by substantial improvements in quality of life and work productivity, consistent with published data showing a clear association between disease severity and impact on health-related quality of life and daily activities.2,23 The improvement in the symptom burden and general wellbeing dimensions on the SHS in the M2x500 group was higher than in the M1000 group, but it should be noted that the lower baseline values in the M1000 group could have skewed results. Furthermore, the improvement in CAI, which includes the leading symptoms of UC such as stool frequency and bloody stools, was similar between the treatment groups and was thus not consistent with the difference observed here.
The efficacy of mesalazine is known to be dose-dependent in both acute 5 and quiescent 24 UC. Randomised dose-finding studies using the Salofalk formulation have reported a dose of 3.0 g/day to be optimal both for achieving clinical remission in UC20,25 and for maintaining remission. 26 Here, 3 g/day delivered either as three 1000 mg tablets or six 500 mg tablets per day resulted in good efficacy outcomes.
Almost half the study population (47.7%) expressed a preference for taking a single, larger high-dose 1000 mg mesalazine tablet compared to two of the smaller conventional 500 mg tablets, while only 10.5% preferred two smaller 500 mg tablets. Fewer tablets could be expected to support treatment adherence.8–10 Poor adherence to the prescribed regimen increases the risk of relapse in quiescent UC27,28 with poorer long-term prognosis. 29 Adherence during short-term treatment of acute flares is relatively high, 5 as might be expected, making an effect on outcomes difficult to establish. However, the preferences expressed here during acute episodes could translate to improved long-term adherence during extended periods of remission, with the potential to reduce relapse rates. Adherence may be further improved by a once-daily mesalazine regimen. Once-daily treatment with mesalazine has been shown to be at least as effective and well tolerated as dosing twice or three times a day for various mesalazine formulations in terms of inducing remission in patients with mild to moderately active UC.20,30,31 As once-daily treatment was not compared with a three times daily regimen in this study, further studies should investigate whether an optimised dose regimen will work with the novel high-dose mesalazine tablet.
No new safety signal was observed for the high-dose 1000 mg mesalazine tablet. The incidence and type of adverse events were similar to the control arm. Oral treatment with either the 1000 mg mesalazine tablet or the conventional regimen was well tolerated, with a low rate of discontinuations due to adverse events (1%).
In conclusion, a novel high-dose 1000 mg mesalazine tablet given three times a day was non-inferior to the registered 500 mg mesalazine tablet for the induction of clinical remission in mild to moderately active UC. The high rates of clinical and endoscopic remission or improvement observed in both treatment arms confirm that oral mesalazine is a powerful and safe first-line treatment modality in patients with mild to moderately active UC. Future studies should explore once-daily dosing with three 1000 mg mesalazine tablets in acute or quiescent UC to optimise further the dosing regimen.
Footnotes
Author contribution
Axel Dignass, Karin Dilger, Roland Greinwald and Tanju Nacak developed the study concept and design. Axel Dignass, Robert Schnabel, Jacek Romatowski, Vladimir Pavlenko, Andrey Dorofeyev, Jelena Derova and Laimas Jonaitis recruited patients to the study and collected data. Axel Dignass, Karin Dilger, Roland Greinwald and Tanju Nacak analysed the data and contributed to writing the manuscript. All authors provided the critical revision of the manuscript for important intellectual content and approved the final version of the manuscript.
Acknowledgements
An initial draft of the manuscript was written by a medical writer (C Dunstall) with funding from Dr Falk Pharma GmbH, Freiburg, Germany. The draft was then extensively reviewed and amended by the authors before submission for publication
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Axel Dignass has received fees and non-financial support from Abbvie, Dr Falk, Ferring, MSD, Takeda, Pharmacosmos, Mundipharma, Vifor, Hospira, Hexal, Allergosan, Janssen, Otsuka and TiGenix. Jacek Romatowski has received non-financial support from Boston Scientific. Karin Dilger, Tanju Nacak and Roland Greinwald are employees of Dr Falk Pharma GmbH. The remaining authors have no conflicts of interest to declare.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the study was funded by Dr Falk GmbH, Freiburg, Germany.