
Abstract
Background
In patients with mesalazine-refractory ulcerative colitis, systemic corticosteroids are the treatment of choice.
Objective
To evaluate the efficacy and safety of prolonged release budesonide granules for the induction of remission in patients with mesalazine-refractory ulcerative colitis.
Methods
Patients with mesalazine-refractory ulcerative colitis discontinued mesalazine at baseline and received 9 mg prolonged release budesonide granules daily for 8 weeks in this open-label, phase IIa study, followed by a 2-week follow-up phase wherein patients continued treatment on alternate days (EudraCT number 2014-005635-14; ClinicalTrials.gov identifier NCT02550418). The primary endpoint was clinical remission (Clinical Activity Index ≤4; stool frequency <18 per week; absence of rectal bleeding) at Week 8. Secondary endpoints included clinical, endoscopic and histological measures of disease at Week 8. A post hoc analysis assessed histo-endoscopic mucosal healing. Treatment-emergent adverse events and morning cortisol levels were assessed throughout the treatment and follow-up phases.
Results
A total of 61 patients were included in the intention-to-treat population; 50 were included in the follow-up analysis set. Clinical remission was achieved in 29 patients (47.5%; 95% confidence interval: 34.6–60.7%) by Week 8. Mean stool and bloody stool frequency decreased significantly from 32.5 to 22.9 per week (p<0.0001) and from 17.6 to 8.1 per week (p<0.0001), respectively. Rates of mucosal healing, endoscopic remission and histological remission were 58.0%, 54.0% and 36.0%, respectively. Histo-endoscopic mucosal healing was achieved by 34.0% of patients. Twenty-four patients (39.3%) experienced treatment-emergent adverse events, of which gastrointestinal disorders (16.4%) were the most common. Mean morning cortisol levels were not significantly suppressed by Week 8.
Conclusions
Treatment with prolonged release budesonide granules for 8 weeks was associated with clinical, endoscopic and histological remission and demonstrated a favourable safety profile in patients with mesalazine-refractory ulcerative colitis. These results warrant further investigation into the potential of prolonged release budesonide granules as an alternative treatment for this patient population.
Key summary
Current knowledge
In patients with ulcerative colitis who are intolerant or refractory to mesalazine, systemic corticosteroids are an effective second-line treatment option, but are associated with well-characterised toxicities Topical corticosteroids such as budesonide have a broad-spectrum action profile, limited systemic bioavailability and less toxicity compared with systemic corticosteroids The current oral formulation with topical corticosteroids may have a suboptimal pH profile for drug release There is a medical need for a topical corticosteroid treatment with a favourable safety profile and an optimised drug release mechanism as an alternative to systemic corticosteroids for patients with mesalazine-refractory ulcerative colitis
What is new?
In this phase IIa study, a novel oral formulation of budesonide granules with a gastro-resistant prolonged release profile was used to target colonic inflammation The novel budesonide formulation was associated with clinical, endoscopic and histological remission in a substantial proportion of patients with mesalazine-refractory ulcerative colitis, with a consistent drug effect throughout the colon The safety profile of prolonged release budesonide was favourable with minimal systemic corticosteroid-associated toxicities These study results warrant further investigation in larger, randomised, controlled clinical studies in patients with mesalazine-refractory ulcerative colitis
Introduction
Ulcerative colitis (UC) is a chronic relapsing/remitting inflammatory bowel disease characterised by the hallmark symptom of bloody diarrhoea due to diffuse inflammation of the colon and rectal mucosa.1
Rectal and oral mesalazine (5-aminosalicylic acid) preparations serve as first-line therapy for active mild-to-moderate UC. For patients intolerant or refractory to mesalazine, systemic corticosteroids, such as prednisolone or equivalents, are an effective second-line treatment option, but their use is limited owing to their well-characterised adverse effects (e.g. Cushing’s syndrome, osteoporosis, osteonecrosis, hypertension, diabetes mellitus, cataract and depression).2,3 Therefore, there is an unmet medical need for novel treatments with a manageable safety profile for patients with mesalazine-refractory UC.
Oral budesonide is a topically active synthetic glucocorticosteroid with a broad-spectrum action profile, limited systemic bioavailability and less corticosteroid-associated toxicities than other corticosteroids.4,5 Different release formulations of budesonide have been optimised for distinct disease patterns of inflammatory bowel diseases, but budesonide MMX® (Multi-Matrix System; a colonic release system) is currently the only approved oral budesonide formulation for the treatment of mild-to-moderate UC.6–8 Budesonide MMX® is released at pH >7,9 which may not be optimal as some healthy subjects may not reach pH 7 in the distal small intestine.10,11 Furthermore, the duration of time the pH is ≥7 during colon passage is significantly shorter in patients with UC compared with healthy subjects (0.3 h versus 9.3 h, respectively; p = 0.005),12 meaning that patients with UC are at risk of suboptimal release of active budesonide.
Accordingly, a novel oral budesonide formulation with a gastro-resistant prolonged release profile starting at pH 6 was developed to target colonic inflammation in patients with UC who have reduced pH values. The drug release mechanism of this novel formulation comprises a solid capsule with a multiparticulate drug delivery system (prolonged release budesonide granules) that facilitates the homogeneous distribution of the drug throughout the colon (Supplementary Figure 1A and B). In Supplementary Figure 2, budesonide plasma profiles for prolonged release budesonide granules and budesonide gastro-resistant capsules (Budenofalk® 3 mg) are included, the latter serving as an oral reference formulation. The prolonged release budesonide granules show a flat plasma profile with late tmax consistent with the desired prolonged release in the colon accompanied with only limited systemic absorption.
Here, we present an open-label, proof-of-concept, phase IIa study of a novel oral formulation of budesonide, as an alternative to systemic corticosteroids, to target colonic regions for the topical treatment of patients with mesalazine-refractory UC.
Methods
Study design
This was an open-label, multicentre, proof-of-concept phase IIa study (TOPICAL-1; EudraCT number 2014-005635-14; ClinicalTrials.gov identifier NCT02550418) conducted between October 2015 and January 2017 at 12 sites in Germany (2), Hungary (4), Latvia (4) and Lithuania (2) (Supplementary Table 1). The study comprised a screening phase of 7–10 days prior to an 8-week, open-label treatment phase, followed by a 2-week follow-up (FU) phase to taper the study drug. Study visits took place at screening (Week −2 to −1), baseline (Week 0) and Weeks 2, 4, 6 and 8 (or withdrawal visit, if earlier). At the screening and Week 8 visits, an endoscopy was performed and the Endoscopic Index (EI) score, Histological Index (HI) score and modified Disease Activity Index (mDAI) subscore were calculated. The last study visit occurred at Week 10 after the 2-week FU phase for tapering of study drug and safety assessments. This study was conducted in accordance with guidelines from the European Medicines Agency for the development of new medicinal products for the treatment of UC.10
Ethics
The study was conducted in accordance with the principles of Good Clinical Practice, the 1975 Declaration of Helsinki, and all applicable national laws and regulations, following approval by competent authorities and independent ethics committees for all participating centres (Supplementary Table 2). Prior to study initiation, written informed consent was obtained from all patients and relevant study data were recorded and analysed pseudonymously.
Eligibility
Patients aged 18–75 years were eligible to take part in the study if they had endoscopically and histologically confirmed active UC, with a Clinical Activity Index (CAI) >4 or ≤12 and EI ≥4, and if the extent of disease was >15 cm above the anus and if the patient had demonstrated an insufficient response or intolerance to treatment with mesalazine (previous or current). It was at the discretion of each investigator to determine whether a patient had experienced an insufficient response or intolerance to treatment with mesalazine (previous or current; oral and/or rectal). Key exclusion criteria were Crohn’s disease, other forms of colitis, coeliac disease, malabsorption syndromes, infections causing bowel disease, abnormal renal and liver function, continuous therapy with CYP3A inducers or inhibitors within 3 weeks prior to baseline, and treatment with immunosuppressants, tumour necrosis factor-α antagonists or anti-integrin therapy within 3 months prior to baseline and/or corticosteroids (oral, inhalative, intravenous or rectal) within 4 weeks prior to baseline.
Study drug and concomitant medication
Patients received one capsule containing 9 mg budesonide gastro-resistant prolonged release granules (provided by Dr Falk Pharma GmbH, Freiburg, Germany) each day for 8 weeks. During this treatment phase, the use of other corticosteroids, immunosuppressants, antidiarrhoeals or antibiotics (except for up to a 7-day course for conditions unrelated to UC) was not permitted. To ensure that prolonged release budesonide granules could be studied without needing to consider the synergistic anti-inflammatory effects of concomitant mesalazine, any mesalazine-containing or -releasing drugs were discontinued at baseline and prohibited during the initial 8-week treatment phase. The 8-week treatment phase was followed by a 2-week FU, wherein patients received one 9 mg budesonide gastro-resistant prolonged release capsule on alternate days.
Study endpoints
Primary endpoint
The primary efficacy endpoint was clinical remission (defined as CAI ≤4, with stool frequency <18 per week and absence of rectal bleeding) at Week 8. CAI was calculated as the sum of the scores of seven variables (number of weekly stools, bloody stools, abdominal pain, general wellbeing, body temperature, extra-intestinal manifestations and erythrocyte sedimentation rate/haemoglobin; Supplementary Table 3).13 The scores for number of weekly stools, bloody stools, abdominal pain and general wellbeing were based on data collected in the patient’s daily diary during the 7 days preceding a study visit. A subgroup analysis of the primary endpoint stratified by localisation of disease (proctosigmoiditis, left-sided or sub-total colitis) was also conducted.
Secondary endpoints
Secondary efficacy endpoints included the following: clinical improvement (defined as a CAI decrease of ≥3 from baseline to Week 8); change in CAI score from baseline to Week 8; clinical remission (defined as an mDAI stool frequency subscore of ≤1 and a rectal bleeding subscore of 0 at Week 8; Supplementary Table 4); change in number of stools and bloody stools per week from baseline to Week 8; time to first resolution of clinical symptoms (defined as the first day of ≥3 consecutive days with either ≤3 stools per day, no bloody stools or ≤3 stools per day without blood); endoscopic improvement (defined as an EI score decrease of ≥1 from baseline to Week 8); endoscopic remission (defined as an EI score of <4; Supplementary Table 5);13 mucosal healing (defined as an mDAI mucosal appearance subscore of ≤1 point, if associated with a decrease of ≥1 point from baseline; any mucosal friability was scored using a mucosal appearance subscore of ≥2);14,15 mucosal improvement (defined as a decrease in the mDAI mucosal appearance subscore of ≥1) at Week 8; combined clinical and endoscopic remission (defined as an mDAI stool frequency subscore and mucosal appearance subscore of ≤1 and an mDAI rectal bleeding subscore of 0); combined clinical and endoscopic remission using a strict definition according to a recent study (defined as an mDAI total score of ≤2 with mDAI stool frequency and rectal bleeding subscores of 0, and mDAI mucosal appearance subscores and mDAI physician’s rating of disease activity subscores of ≤1);8 histological remission (defined as an HI of ≤1, which signifies a complete absence of neutrophils in the lamina propria and epithelium, no crypt abscesses, no mucin depletion, normal surface epithelial integrity, no or mild round cells in the lamina propria or epithelium, mild-to-moderate crypt architectural irregularities and no erosions or ulcers; Supplementary Table 6); histological improvement (defined as a decreased HI of ≥1 from baseline scores of 2, 3 or 4 at Week 8)16 and change in the number of patients with faecal calprotectin ≤50 μg/g and between >50 to ≤250 μg/g from baseline to Week 8. Subgroup analyses of mucosal healing and histological remission stratified by localisation of disease (proctosigmoiditis, left-sided or subtotal/total colitis) were also conducted.
Post hoc analyses
Owing to an updated definition of mucosal healing by the US Food and Drug Administration (FDA),17 requiring a histological and visual assessment of the mucosa, a post hoc analysis was conducted to assess histo-endoscopic mucosal healing (defined as an mDAI mucosal appearance score and an HI score of ≤1) at Week 8.
Safety
For safety analyses, treatment-emergent adverse events (TEAEs), as defined by MedDRA v19.1, and mean morning cortisol levels were recorded throughout the treatment and FU phases.
Statistical analysis
Primary efficacy and secondary efficacy endpoints were analysed by descriptive statistics of the intention-to-treat (ITT) population. The ITT population comprised all patients who received at least one dose of study drug. The FU analysis set comprised all patients who received at least one dose of study drug during the FU phase and had at least one FU value for the safety endpoints to be analysed. The safety analysis set (SAF) comprised all patients who received at least one dose of study drug and had at least one FU value for the safety variables to be analysed. The SAF was used for all analyses of safety parameters. The sample size calculation assumed a primary endpoint clinical remission rate of 40% based on a recent study.18 The two-sided 95% confidence interval (CI) for the remission rate has a width of 25% if 60 patients were evaluated. Clinical endpoints and morning cortisol levels were analysed using last observation carried forward.
Results
Study population
Overall, 66 patients were screened, and 61 patients received prolonged release budesonide granules and were included in the ITT population and the SAF. Fifty patients qualified for inclusion in the FU analysis set (Figure 1).
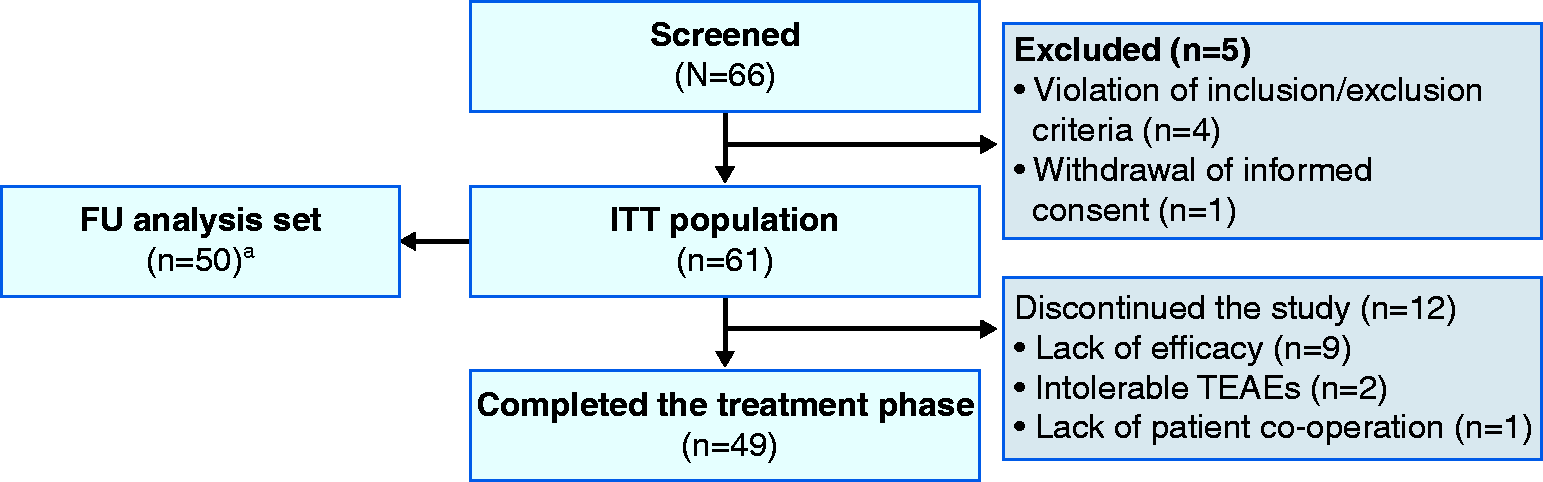
Patient disposition.
Patient demographics and baseline characteristics are summarised in Table 1. Most patients (n = 59, 96.7%) had an insufficient response or intolerance to treatment with mesalazine (previous or current) prior to baseline as determined by the investigators. The median duration of mesalazine treatment prior to baseline was 57.5 days (interquartile range (IQR): 28.0, 139.0). During this treatment, patients received a median maximum oral daily dose of 3.0 g (IQR: 2.4, 3.5) and 10 patients received rectal mesalazine treatment with a median maximum daily dose of 2.5 g (IQR: 1.0, 4.0).
Patient demographics and baseline characteristics (ITT population).
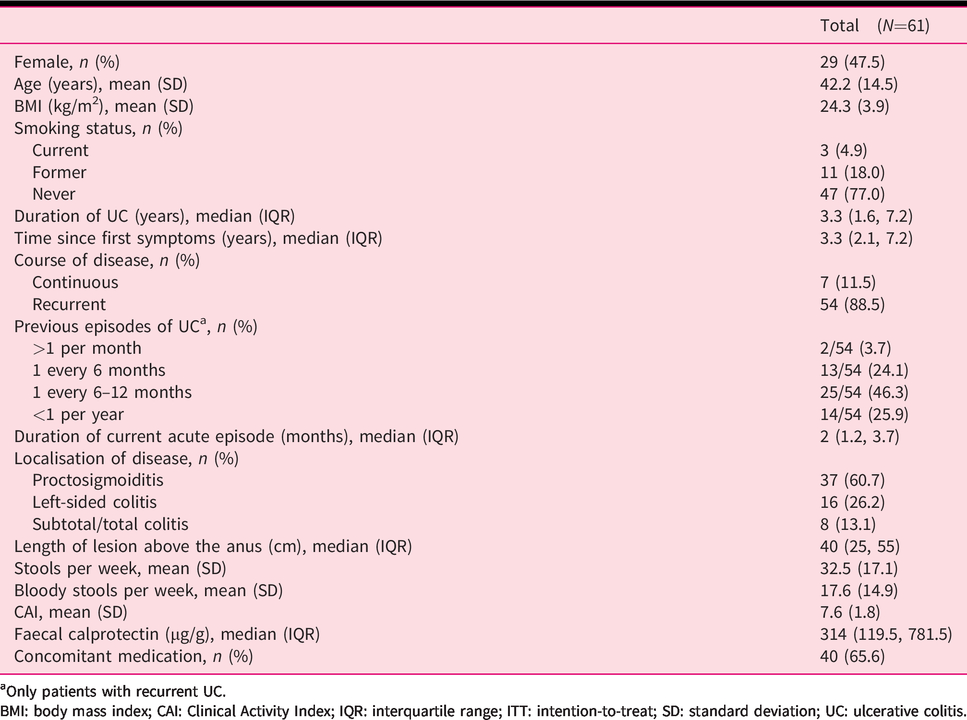
aOnly patients with recurrent UC.
BMI: body mass index; CAI: Clinical Activity Index; IQR: interquartile range; ITT: intention-to-treat; SD: standard deviation; UC: ulcerative colitis.
Treatment compliance
Overall, 60/61 patients (98.4%) administered ≥80% of the study drug during the treatment phase, which corresponded to a mean compliance rate of 97.9%. During the FU phase, 48/50 patients (96.0%) administered ≥80% of the study drug, which corresponded to a mean compliance rate of 116.3%.
Primary efficacy endpoint
The primary efficacy endpoint of clinical remission (defined as CAI ≤4, with stool frequency <18 per week and absence of rectal bleeding) was achieved by 29/61 patients (47.5%; 95% CI: 34.6–60.7%) (Figure 2). In subgroup analyses of the primary efficacy endpoint, clinical remission rates were greater in patients with subtotal/total colitis (62.5%) compared with patients with proctosigmoiditis (45.9%) or left-sided colitis (43.8%). The primary efficacy endpoint was achieved by 15/33 patients with mild disease activity (CAI ≤7 at baseline; 45.5%; 95% CI: 28.1–63.7%) and by 14/28 patients with moderate disease activity (CAI >7 at baseline; 50.0%; 95% CI: 30.7–69.4%).
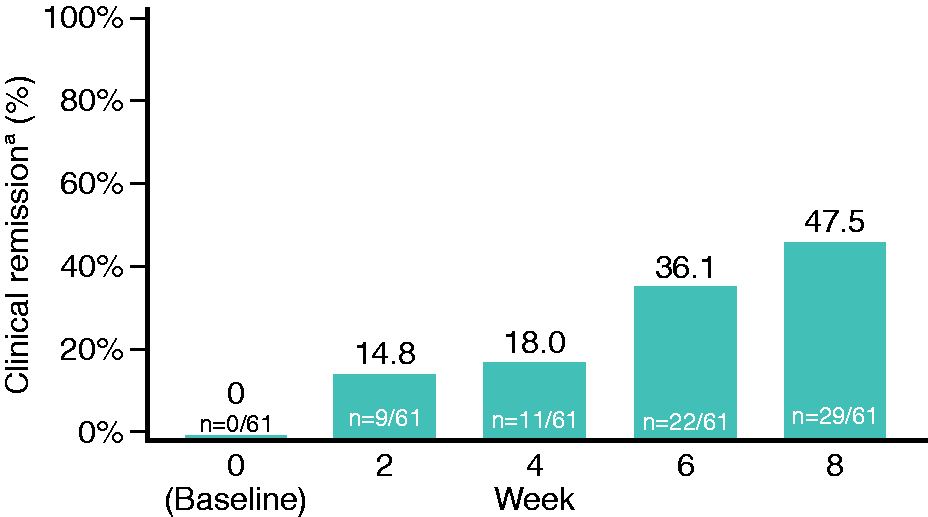
Primary endpoint: clinical remission.
Secondary endpoints and post hoc analyses
Clinical assessments
By Week 8, clinical improvement was achieved by 37/61 patients (60.7%) and clinical remission (defined as mDAI stool frequency subscore of ≤1 and rectal bleeding subscore of 0 at Week 8) was achieved by 35/61 patients (57.4%) (Table 2). Mean CAI score, stools per week and bloody stools per week significantly decreased from baseline to Week 8 (p<0.0001; Table 2). The median time to first resolution of symptoms (defined as the first day of at least ≥3 consecutive days with either ≤3 stools per day, no bloody stools per day or ≤3 stools per day without blood) was 5 days, 21 days and 31 days, respectively. Mean mDAI score significantly decreased from baseline to Week 8 (p<0.0001; Table 3).
Clinical, endoscopic and histological endpoints (ITT population).
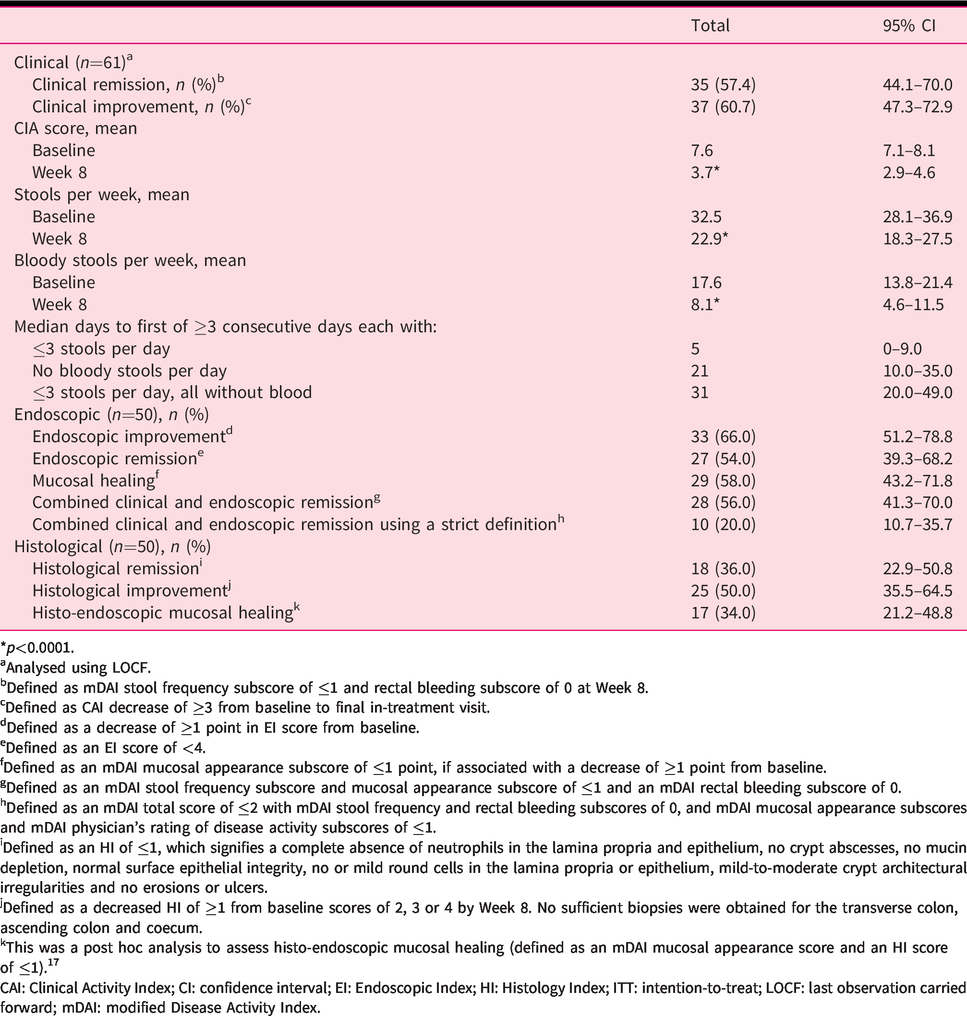
*p<0.0001.
aAnalysed using LOCF.
bDefined as mDAI stool frequency subscore of ≤1 and rectal bleeding subscore of 0 at Week 8.
cDefined as CAI decrease of ≥3 from baseline to final in-treatment visit.
dDefined as a decrease of ≥1 point in EI score from baseline.
eDefined as an EI score of <4.
fDefined as an mDAI mucosal appearance subscore of ≤1 point, if associated with a decrease of ≥1 point from baseline.
gDefined as an mDAI stool frequency subscore and mucosal appearance subscore of ≤1 and an mDAI rectal bleeding subscore of 0.
hDefined as an mDAI total score of ≤2 with mDAI stool frequency and rectal bleeding subscores of 0, and mDAI mucosal appearance subscores and mDAI physician’s rating of disease activity subscores of ≤1.
iDefined as an HI of ≤1, which signifies a complete absence of neutrophils in the lamina propria and epithelium, no crypt abscesses, no mucin depletion, normal surface epithelial integrity, no or mild round cells in the lamina propria or epithelium, mild-to-moderate crypt architectural irregularities and no erosions or ulcers.
jDefined as a decreased HI of ≥1 from baseline scores of 2, 3 or 4 by Week 8. No sufficient biopsies were obtained for the transverse colon, ascending colon and coecum.
kThis was a post hoc analysis to assess histo-endoscopic mucosal healing (defined as an mDAI mucosal appearance score and an HI score of ≤1).17
CAI: Clinical Activity Index; CI: confidence interval; EI: Endoscopic Index; HI: Histology Index; ITT: intention-to-treat; LOCF: last observation carried forward; mDAI: modified Disease Activity Index.
mDAI, EI and HI scores.
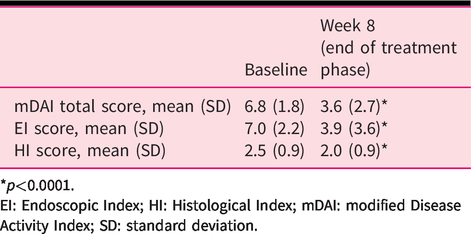
*p<0.0001.
EI: Endoscopic Index; HI: Histological Index; mDAI: modified Disease Activity Index; SD: standard deviation.
Endoscopic assessments
By Week 8, endoscopic improvement was achieved by 33/50 patients (66.0%) and endoscopic remission was achieved by 27/50 patients (54.0%) (Table 2). Mean EI score significantly decreased from baseline to Week 8 (p<0.0001; Table 3). Mucosal healing (defined as an mDAI mucosal appearance subscore of ≤1 point, if associated with a decrease of ≥1 point from baseline) was achieved by 29/50 patients (58.0%) at Week 8. In a subgroup analysis, rates of mucosal healing were greater in patients with subtotal/total colitis (83.3%) than in patients with proctosigmoiditis (55.2%) or left-sided colitis (53.3%; Table 4).
Subgroup analysis of clinical remission, mucosal healing and histological remission stratified by localisation of disease (ITT population).
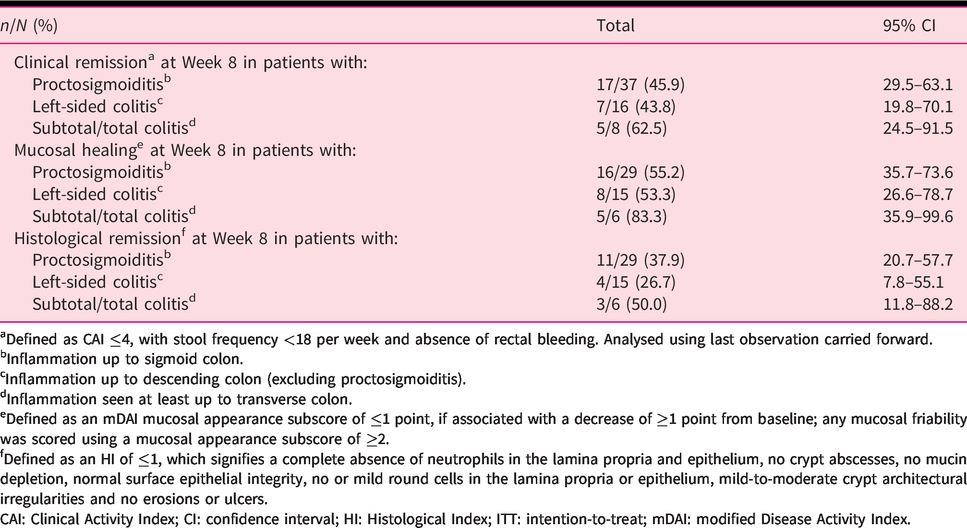
aDefined as CAI ≤4, with stool frequency <18 per week and absence of rectal bleeding. Analysed using last observation carried forward.
bInflammation up to sigmoid colon.
cInflammation up to descending colon (excluding proctosigmoiditis).
dInflammation seen at least up to transverse colon.
eDefined as an mDAI mucosal appearance subscore of ≤1 point, if associated with a decrease of ≥1 point from baseline; any mucosal friability was scored using a mucosal appearance subscore of ≥2.
fDefined as an HI of ≤1, which signifies a complete absence of neutrophils in the lamina propria and epithelium, no crypt abscesses, no mucin depletion, normal surface epithelial integrity, no or mild round cells in the lamina propria or epithelium, mild-to-moderate crypt architectural irregularities and no erosions or ulcers.
CAI: Clinical Activity Index; CI: confidence interval; HI: Histological Index; ITT: intention-to-treat; mDAI: modified Disease Activity Index.
Combined clinical and endoscopic remission was achieved by 28/50 patients (56.0%) and combined clinical and endoscopic remission using a strict definition was achieved by 10/50 patients (20.0%) by Week 8.
Histological assessments
Mean HI score significantly decreased from baseline to Week 8 (p<0.0001; Table 3). Histological improvement was achieved by 25/50 patients (50.0%). Histological remission (including a complete absence of neutrophils in the lamina propria and epithelium, no crypt abscesses, no mucin depletion, normal surface epithelial integrity, no or mild round cells in the lamina propria or epithelium, mild-to-moderate crypt architectural irregularities and no erosions or ulcers) was achieved by 18/50 patients (36.0%) at Week 8 (Table 2). In a subgroup analysis, rates of histological remission were greater in patients with subtotal/total colitis (50.0%) than in patients with proctosigmoiditis (37.9%) or left-sided colitis (26.7%; Table 4).
Recently updated FDA guidance states that both a visual assessment of mucosal appearance and a histological assessment are required.17 Therefore, we conducted a post hoc analysis of histo-endoscopic mucosal healing, which was achieved by 17/50 patients (34.0%).
Faecal calprotectin assessment
The proportion of patients with faecal calprotectin levels of >50 to ≤250 μg/g or >250 μg/g decreased from 17/61 patients (27.9%) and 35/61 patients (57.4%) at baseline to 11/50 patients (22.0%) and 24/50 patients (48.0%) at Week 8, respectively. The proportion of patients with faecal calprotectin levels of ≤50 μg/g increased from 8/61 patients (13.1%) at baseline to 14/50 patients (28.0%) at Week 8.
Safety
Overall, 24 patients (39.3%) experienced a total of 40 TEAEs during the 8-week open-label treatment phase. The most common TEAEs were a flare of the underlying disease (UC) (n = 8, 13.1%), headache (n = 4, 6.6%), large intestinal polyp (n = 2, 3.3%) and respiratory tract infection (n = 2, 3.3%). One patient experienced a serious TEAE (increased activity of UC), which was deemed non-drug related by the investigator. Seven patients (11.5%) experienced drug-related TEAEs (eosinophil count increased, eosinophilia, headache, large intestinal polyp (recorded by the investigator as ‘polypoid changes in the colon mucosa’), nausea, neutropenia and oral mucosal erythema). TEAEs led to treatment discontinuation in eight patients (13.1%), of which aggravation of UC was the most common (n = 6, 9.8%).
During the FU phase, two patients experienced four TEAEs (appendicitis, peritonitis, rash and renal cysts). One patient experienced a serious TEAE (appendicitis) during the FU phase, which was deemed non-drug related by the investigator. One TEAE (rash) was assessed as drug related by the investigator and study sponsor.
Mean (standard deviation) morning cortisol levels remained stable with 11.5 μg/dL (4.5 μg/dL) at baseline and 11.2 μg/dL (4.4 μg/dL) at Week 8, with few patients deviating from the normal range (6.2–18.0 μg/dL; Table 5). During the FU phase, mean morning cortisol levels remained within the normal range.
Cortisol levels.
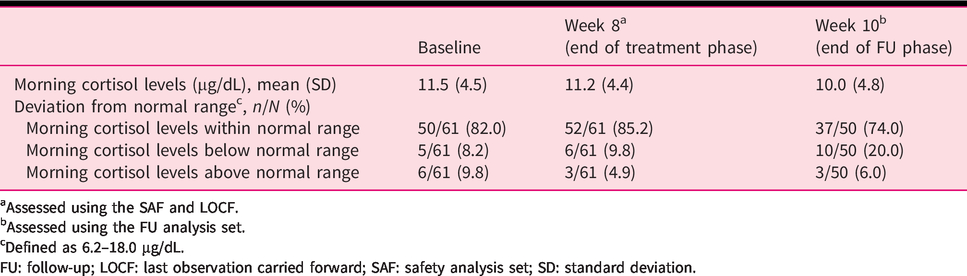
aAssessed using the SAF and LOCF.
bAssessed using the FU analysis set.
cDefined as 6.2–18.0 μg/dL.
FU: follow-up; LOCF: last observation carried forward; SAF: safety analysis set; SD: standard deviation.
Discussion
There is currently a medical need for a topical corticosteroid treatment with a favourable safety profile and an optimised drug release mechanism for patients with mesalazine-refractory mild-to-moderate UC. Budesonide MMX® is a topical corticosteroid that has fewer corticosteroid-associated toxicities compared with systemic corticosteroids but has a pH profile that may be suboptimal for drug release in patients with UC. Therefore, we investigated the efficacy and safety of a novel oral formulation of budesonide granules with a gastro-resistant prolonged release profile for the topical treatment of patients with mesalazine-refractory UC.
In this open-label study, prolonged release budesonide granules showed a consistent drug effect throughout the colon and was associated with clinical, endoscopic and histological remission in a substantial number of patients with mesalazine-refractory UC.
The primary endpoint clinical remission rate of 47.5% demonstrated herein is largely consistent with a prior randomised controlled study, in which patients with active mild-to-moderate UC received 3 g/day mesalazine or 9 mg/day budesonide.18 In that study, mesalazine was superior to budesonide, yet clinical remission was also attained under budesonide in a high proportion of patients (ITT: 39.5%) using the same primary efficacy endpoint (CAI score ≤4, with subscores for stool frequency and rectal bleeding of 0) used in our study.18 Notably, the budesonide formulation used in that study was designed for drug release in the terminal ileum, and hence was not optimised for use in UC. Moreover, the study population in our study was refractory to mesalazine, thus patients may have had a more recalcitrant disease pattern. The difference in clinical remission rates between our study and the prior study may be attributed to the optimised colonic release of budesonide granules.
The combined clinical and endoscopic remission rate, using a strict definition, demonstrated in our open-label phase IIa study was 20.0%. In a recent randomised controlled phase III study of budesonide MMX® versus placebo in patients with UC, the combined clinical and endoscopic remission rate was 13.0%.8 In that study, patients were permitted to receive concomitant oral mesalazine ≥2.4 g/day during the 8-week treatment phase whereas, in our study, mesalazine was prohibited. Therefore, our study provides unique insight into the treatment effect of prolonged release budesonide granules alone, without needing to consider the synergistic anti-inflammatory effects of concomitant mesalazine. Additional strengths of our study are that we used an established dosing regimen, and objective assessments and measured faecal calprotectin levels to investigate the efficacy and safety of prolonged release budesonide. Our study also included a post hoc analysis of histo-endoscopic mucosal healing that was achieved by 17/50 patients (34.0%). Histo-endoscopic mucosal healing is currently regarded by the research community19–22 and regulatory bodies10,17 to be the most complete method of assessing mucosal healing.23
Prolonged release budesonide granules demonstrated a favourable safety profile with minimal systemic corticosteroid-associated toxicities. The most common TEAE was a flare of the underlying disease (UC), likely caused by lack of efficacy in this difficult-to-treat patient population. Few patients experienced TEAEs during FU, suggesting that tapering the dose of budesonide granules is associated with a favourable safety profile. Mean morning cortisol levels remained stable within the normal range during the treatment and FU phases, meaning that a clinically significant suppression of the hypothalamic–pituitary–adrenal axis function was not detected. Therefore, a FU phase for tapering of prolonged release budesonide granules may not be required.
This study was limited by its open-label design and the absence of a control group, which confounded our ability to compare the efficacy and safety of prolonged release budesonide with other treatments for this patient population. The sample size of 61 patients was relatively small and only included patients from four European countries. The mean treatment compliance rate during the FU phase was above 100% (116.3%) because some patients continued to administer one capsule per day during the FU phase instead of one capsule every other day as outlined in the study protocol.
In conclusion, 8-week treatment with prolonged release budesonide granules in the absence of concomitant mesalazine was associated with clinical, endoscopic and histological remission in patients with UC who were intolerant or refractory to mesalazine. These study results warrant further investigation of prolonged release budesonide in larger and randomised controlled clinical studies.
Supplemental Material
sj-pdf-1-ueg-10.1177_2050640620962632 - Supplemental material for Efficacy and safety of prolonged release budesonide granules in mesalazine-refractory ulcerative colitis: A multi-centre Phase IIa study (TOPICAL-1)
Supplemental material, sj-pdf-1-ueg-10.1177_2050640620962632 for Efficacy and safety of prolonged release budesonide granules in mesalazine-refractory ulcerative colitis: A multi-centre Phase IIa study (TOPICAL-1) by Klaus Fellermann, Ingolf Schiefke, István Rácz, Jelena Derova, Laimas Jonaitis, Sarah Wehrum, Tanju Nacak and Roland Greinwald in United European Gastroenterology Journal
Footnotes
Acknowledgements
Medical writing support was provided by Matthew Reynolds from OPEN Health Medical Communications, London, United Kingdom, funded by Dr Falk Pharma GmbH.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KF reports receiving personal fees related to this submitted work for his participation as a principal investigator and for travel expenses from Dr Falk Pharma GmbH. IS, IR, JD and LJ report no conflicts of interest. SW reports her employment at Dr Falk Pharma GmbH. TN and RG report their employment at Dr Falk Pharma GmbH and have a patent pending (EP18190638) that is relevant to the submitted work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Dr Falk Pharma GmbH.
Ethics approval
The study was approved by competent authorities and independent ethics committees for all participating centres.
Informed consent
Prior to study initiation, written informed consent was obtained from all patients and relevant study data were recorded and analysed pseudonymously.
Manuscript note
KF was affiliated with University Hospital Schleswig Holstein at the time of the study and is now affiliated with Medizinische Klinik I, Krankenhäuser Landkreis Freudenstadt gGmbH, Freudenstadt, Germany.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.