
Abstract
Retinoschisin protein is mainly produced in the retina, with minimal expression in other tissues. It is located on the surface of the photoreceptor inner segments and bipolar cells. X-linked retinoschisis, caused by mutations in the RS1 gene, primarily presents as cavities in the retina and separation of the layers. We report a case of a 2-year-old male patient with notable clinical differences in both eyes. During follow-up, genetic testing confirmed an RS1 gene mutation causing retinal detachment and X-linked retinoschisis, with no family history of the disease. The clinical diagnosis of retinal detachment may seem straightforward, but its cause is complex and diverse. For patients with retinal detachment, clinicians need to determine its underlying cause. It is important to note that patients with X-linked retinoschisis may exhibit different phenotypes in each eye, and genetic testing is essential for the clinical diagnosis of X-linked retinoschisis.
Introduction
Retinoschisin (RS1) protein is a secreted protein that plays a crucial role in maintaining retinal stability, and many mutations in the RS1 gene are associated with X-linked retinoschisis (XLRS). XLRS is an X-chromosome recessive inherited fundus disorder that is most common in males and typically results in bilateral vision impairment. Its characteristic clinical features include a microcystic spoke-wheel pattern and foveal schisis caused by retinoschisis, which affects the inner retinal layers and often results in moderate to severe central vision loss. Complications such as retinal detachment (RD) and vitreous hemorrhage mainly occur during the juvenile period.1,2 The RS1 gene, located in the Xp22 region of the short arm of the X chromosome, consists of six exons and five introns covering 32.43 kb of genomic DNA. This mRNA is translated into a protein of 224 amino acids, known as the RS1 protein. 3 How the RS1 protein maintains retinal stability remains unclear, despite recent studies revealing its molecular structure. We identified a splice site mutation in the RS1 gene in a Chinese patient with RD. The proband showed significant asymmetry in both eyes, providing a reference for diagnosing XLRS. For patients with RD, clinicians need to determine its cause. In this article, we present a case report of this patient and discuss the relationship between the RS1 gene mutation and the development of RD, as well as its role in maintaining the structural integrity of the retina.
Case presentation
Clinical data
The proband was a 2-year-old boy whose parents noticed exotropia in his right eye and brought him to our hospital. Initial visual acuity was finger counting at face in the right eye and finger counting at one meter in the left eye. Intraocular pressure measured by Goldmann applanation tonometry was 12 mmHg in the right eye and 13 mmHg in the left eye. B-scan ultrasound revealed a “V”-shaped, highly echogenic band in the vitreous cavity of the right eye, with its apex attached to the optic disc, while the left eye showed a strip-shaped, highly echogenic band in the vitreous cavity (Figure 1(a) and (b)). A clinical diagnosis of RD in the right eye was made. Subsequently, the proband underwent surgical treatment at another hospital. During follow-up at our institution after the first silicone oil removal, visual acuity in the right eye was counting fingers, and intraocular pressure was 15 mmHg. Scanning laser ophthalmoscopy (SLO) showed residual gas and epiretinal proliferation in the right vitreous cavity, causing retinal traction (Figure 1(c)), but optical coherence tomography (OCT) of the right eye could not be visualized. Due to epiretinal proliferation, the patient underwent another surgery at that hospital. After the second silicone oil removal, visual acuity was light perception in the right eye, and intraocular pressure was measured at 10 mmHg in the same eye. Follow-up at our hospital showed pupil occlusion with posterior adhesion to the iris and turbidity of the refractive stroma in the right eye (Figure 1(d)). B-scan ultrasound showed a point and patchy, strong echogenic bands in the right vitreous cavity (Figure 1(e)). SLO of the left eye showed epiretinal proliferation (Figure 1(f)), while OCT revealed thickened foveal retina, cystoid hyporeflective spaces in the inner and outer nuclear layers, and bridged hyporeflective spaces in the peripheral retinal layers (Figure 1(g) and (h)). Thus, the clinical diagnosis for the left eye was retinoschisis.
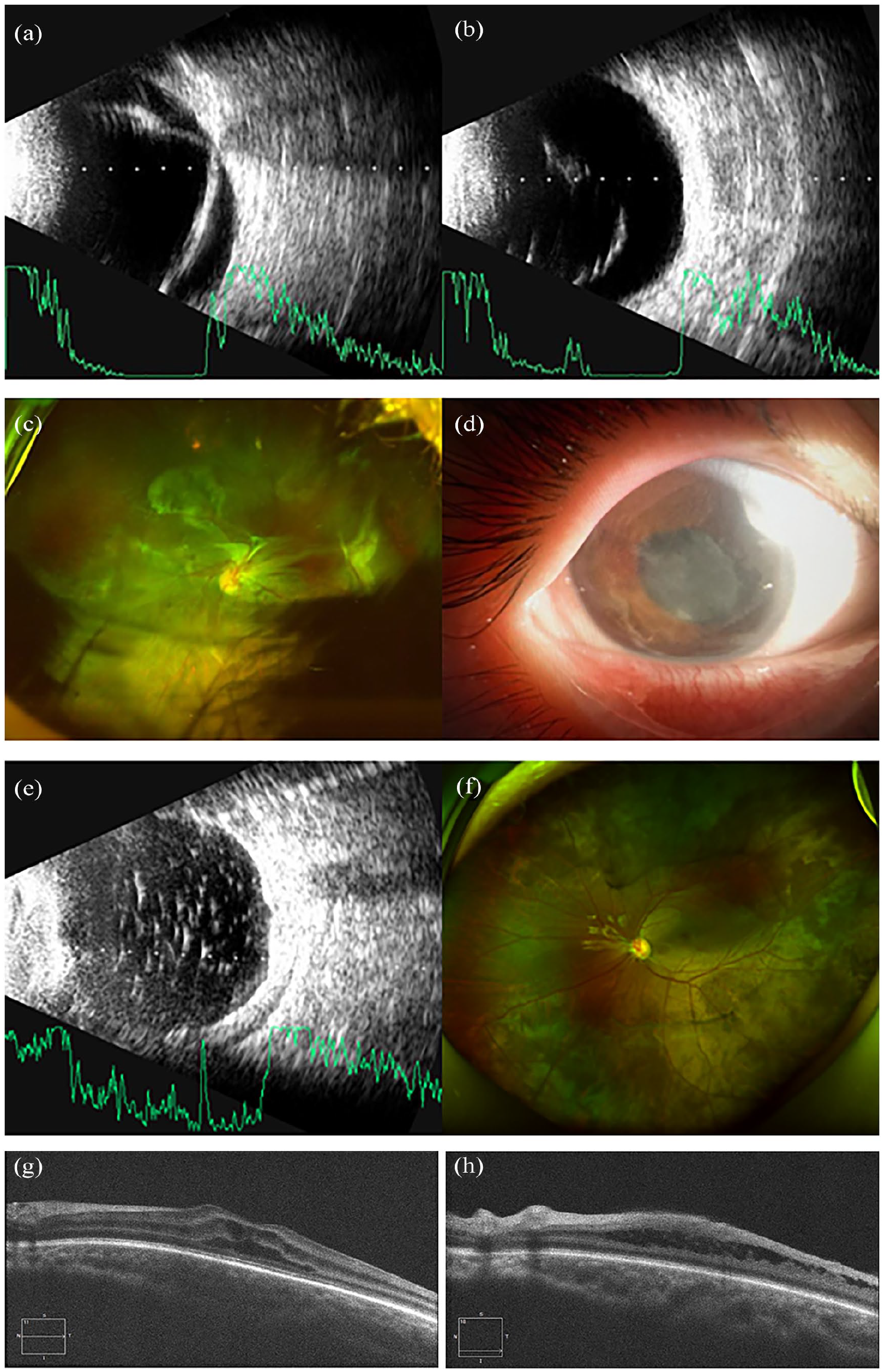
Eye examinations of the proband. B-scan images of the right eye (a) and left eye (b). SLO after the first silicone oil removal surgery of the right eye (c). Anterior segment photograph (d) and B-scan (e) images after the second silicone oil removal surgery of the right eye. SLO (f) and OCT (g, h) images of the left eye.
During this period, all four of the proband’s surgeries were carried out at an external hospital. The proband was initially diagnosed there with rhegmatogenous RD (RRD) in the right eye and underwent pars plana vitrectomy, lensectomy, and silicone oil tamponade. During the procedure, a total RD was observed, along with a horseshoe-shaped retinal tear in the superotemporal quadrant, and postoperative topical treatment with glucocorticoids, Nonsteroidal Antiinflammatory Drugs (NSAIDs), and mydriatics were used to prevent proliferative vitreoretinopathy. Silicone oil removal took place 4 months after the initial surgery. Seven months after the first oil removal, the proband received additional silicone oil tamponade due to traction from epiretinal proliferation, followed by a second oil removal 4 months later. Fundus examination of the proband’s parents showed no relevant clinical phenotypes. Whole-exome sequencing of all known retinal disease-related genes was performed on the proband and his parents.
Genetic mutation analysis results
DNA was extracted from peripheral blood following standard procedures. Panel sequencing identified the RS1 gene splice site mutation c.53-1G>A. A literature review revealed a case report linking this RS1 gene mutation site to XLRS. 4 Sanger sequencing was used for validation, showing that the proband had a hemizygous mutation c.53-1G>A in the RS1 gene (Figure 2(a)). His mother was a heterozygous carrier of the mutation, while his father showed no mutation (Figure 2(b) and (c)). This suggests the mutation was inherited from the mother, whose fundus examination showed no related clinical phenotypes. His mother was healthy, consistent with family co-segregation. According to the American College of Medical Genetics and Genomics guidelines, 5 the mutation was classified as pathogenic (PVS1 + PP3 + PS4 + PM2).

Genetic sequencing of the proband and his parents. The proband (a) shows the RS1 gene splice mutation site c.53-1G>A (red arrow). The proband’s mother (b) shows heterozygous mutation in the RS1 gene c.53-1G>A (red arrow). The proband’s father (c) shows no mutation at this site (red arrow).
Discussion
In this study, we report a case of XLRS with RD caused by a pathogenic mutation in the RS1 gene. The proband showed significant heterogeneity in clinical phenotypes in both eyes, with the left eye showing fovea schisis and peripheral retinoschisis, while the right eye had RD. Although Wang et al. 4 recently reported that this site is associated with XLRS, all patients showed typical foveal schisis in both eyes, and none of them developed RD.
XLRS primarily affects both retinas and shows high genetic and phenotypic diversity. 6 Retinoschisis mainly occurs in the inner layer of the retina. OCT shows retinal cystoid spaces in different layers, mostly in the inner nuclear layer, with bridge-like connections separating small cystoid spaces from each other.7,8 About 50% of XLRS patients have peripheral retinoschisis, which is more common in those with early-onset XLRS.6,7 RD occurs in 5%–22% of cases, usually as RRD caused by fractures in the inner and outer cystoid walls of peripheral retinoschisis.7,9 In addition, recurrent vitreous hemorrhage accumulation can lead to the formation of a proliferative membrane on the retina, pulling on the posterior vitreous cortex, and causing RD. 10 The younger the child, the higher the likelihood of peripheral retinoschisis, which can lead to complications such as vitreous hemorrhage and RD.6,7,9,11 In some patients under 2 years old, XLRS often shows with strabismus or nystagmus. Severe retinoschisis, vitreous hemorrhage, and RD can result in varying degrees of decreased visual acuity.12–14 In this study, although the proband was in the juvenile period, panretinal detachment had already occurred in the right eye at the first visit, and we could not determine whether the RD was caused by retinoschisis. The fundus appearance showed significant asymmetry in both eyes, whereas previous studies commonly report symmetrical fundus appearances in both eyes. 15 Therefore, it is important to recognize that patients with XLRS may exhibit different phenotypes in each eye, although the reason remains unknown.
This splice site mutation may create an alternative splice acceptor or lead to exon 2 skipping, 4 resulting in abnormally spliced mRNA that affects the RS1 protein. The RS1 protein consists of four parts: a cleavable N-terminal signal sequence, an RS1 domain, a highly conserved discoidal domain, and a C-terminal segment. This protein forms a complex homo-oligomeric structure that strongly binds to photoreceptors and bipolar cells; it resembles many other adhesion-related proteins and plays a crucial role in the structural organization of the retina.3,16 Moreover, the RS1 protein might function as part of the retinal intercellular matrix, interacting with numerous proteins such as NaK-ATPase, and help regulate the intracellular and extracellular environments, especially the fluid balance between photoreceptors and bipolar cells.3,17,18 Its unique structural features are essential for cell adhesion, cell-cell interactions, and maintaining the integrity of the retinal structure. In addition, Huang et al. reported numerous gene expression changes suggesting the RS1 protein has a direct or indirect role in retinal development. 19
Furthermore, Gene Ontology (GO) enrichment analysis in Homo sapiens showed that enriched biological processes included retina layer formation and retina morphogenesis in camera-type eyes (Figure 3). The RS1 gene splice mutation c.53-1G>A caused abnormal changes in the RS1 protein, which may have disrupted the tight connection between photoreceptors and bipolar cells or affected the development and structure of retinal cells and tissues. In other words, the RD in the proband’s right eye could have resulted from retinoschisis, or the abnormal change in the RS1 protein might have impacted retinal structural integrity and development, leading to RD.

GO enrichment analysis of the RS1 gene (Homo sapiens): BP, CC, and MF. The color indicates the p value size.
The clinical signs of XLRS are nonspecific, so diagnosis relies on fundus photography, OCT, electroretinography, and genetic testing. The proband did not undergo an electroretinogram, so we could not compare results with genotypes. In addition, we were unable to determine whether the RD in the proband’s right eye resulted from peripheral retinoschisis related to XLRS or from abnormal changes in the RS1 protein. There is still no clear understanding of how the RS1 protein supports the interlayer structure of the retina. The role of the RS1 protein in RD development remains to be investigated.
Conclusion
Therefore, further research is needed to clarify the specific nature of this splice site mutation and its connection to fundus phenotypic heterogeneity. Ultimately, in the future, patients with this particular type of RD should undergo as comprehensive an examination as possible. If necessary, genetic testing should be performed to aid in diagnosis and understanding of the cause, which will help ensure timely and effective intervention and treatment.
Footnotes
Ethical Considerations
Ethical approval was provided by the Ethics Committee of the Affiliated Hospital of Zunyi Medical University (no. KLLY-2021-131).
Consent for Publication
Written informed consent was obtained from the patient’s parents for the publication of this case report.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Natural Science Foundation of China (no. 31871261), the Guizhou Province Basic Research Program Project (Guizhou Science and Technology Cooperation Foundation under Grant ZK (2021) general 423; Guizhou Science and Technology Cooperation Foundation under Grant ZK (2021) general 428; Guizhou Science and Technology Cooperation Foundation under Grant ZK (2022) general 647; Guizhou Science and Technology Cooperation Foundation under Grant ZK (2023) general 529), the Guizhou Province Science and Technology Support Program Project (Guizhou Science and Technology Cooperation Support (2023) general 265) and the Science and Technology Program of Guizhou Province, Basic Research of Qiankehe (No. ZK[2023]-529)
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.