
Abstract
Sarcomas are a heterogeneous group of mesenchymal tumours that can affect the skin. They are dominated by Kaposi’s sarcoma and dermatofibrosarcoma. Cutaneous leiomyosarcoma is a very rare entity. We report the case of a 78-year-old patient who presented with a primary frontal cutaneous leiomyosarcoma treated surgically with a good outcome at the last follow-up. The objective of this work is to identify the epidemiological, clinical, anatomopathological, therapeutic and prognostic particularities of this pathology through a review of the literature.
Introduction
Cutaneous leiomyosarcoma (cLMS) is a very rare subtype of sarcomas affecting the skin. It accounts for 2%–3% of all cutaneous soft tissue sarcomas, far behind Kaposi’s sarcoma (71%) and dermatofibrosarcoma (18%).1–4 Histologically, cLMS is a primary skin tumour, arising from normal arrector pili muscle. On immunohistochemical study, this tumour classically exhibits positive reaction with desmin, smooth muscle actin and H-caldesmone. Its aetiology remains unknown. Some rare cases have been reported in the literature notably in Li-Fraumeni Syndrome or in association with Epstein-Barr virus in immunocompromised patients.2,5,6
Typically, cLMS presents as a painless nodule located on the trunk or the lower limbs. It mostly affects older Caucasian males with a peak incidence between the fifth and seventh decades of life.2,7 Exceptionally, paediatric forms have been reported, occurring in immunocompromised patients.5,6 Compared to other primary leiomyosarcoma (LMS) (soft tissue and uterine), cLMSs have a more favourable prognosis, especially when the lesion is confined to the dermis.
Due to the rarity of this entity, data are still limited and mostly from single-centre retrospective studies with a small sample size.
This case report and review of the literature aims to study the epidemiological, clinical, anatomopathological, therapeutic and prognostic aspects of this pathology.
Case report
A 78-year-old patient, with a history of chronic obstructive pulmonary disease, presented to the outpatient clinic complaining of a left temporal ulcerated mass. The lesion appeared 3 months ago and was reportedly rapidly increasing in size. The patient did not recall any trauma or septic inoculation. Furthermore, he did not report any family history of neoplasms. On physical examination, there was a 5-cm nodular and ulcerated mass with a wide implantation base, located in the left frontotemporal region (Figure 1). The movable mass was firm and painless to touch. The overlying skin was ulcerated and surrounded by inflammatory reaction. The remainder of the physical examination did not reveal any abnormalities, particularly no lymph node enlargement. Commuted tomography scan showed thickening of the left frontotemporal subcutaneous soft tissue without extension to the underlying bone.
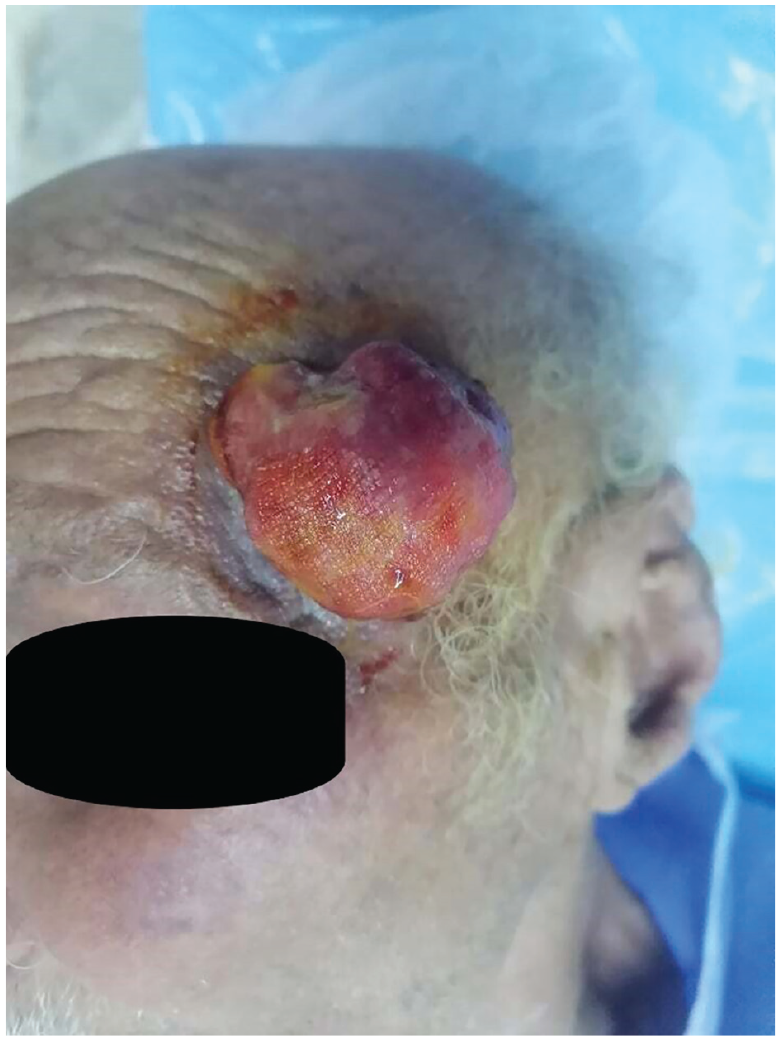
Clinical appearance of the tumour.
A biopsy specimen was sent for pathological examination. Microscopically, the tumour was a malignant mesenchymal proliferation made up of spindle cells arranged in long, intersecting bundles. Tumour cells had enlarged, pleomorphic and hyperchromatic nuclei with atypical mitosis. On immune stains, these cells were highly reactive with smooth muscle markers: H-Caldesmone and smooth muscle actin. CD 34, PS 100, HMB45 and cytokeratin were negative. The diagnosis of a LMS was retained (Figure 2).
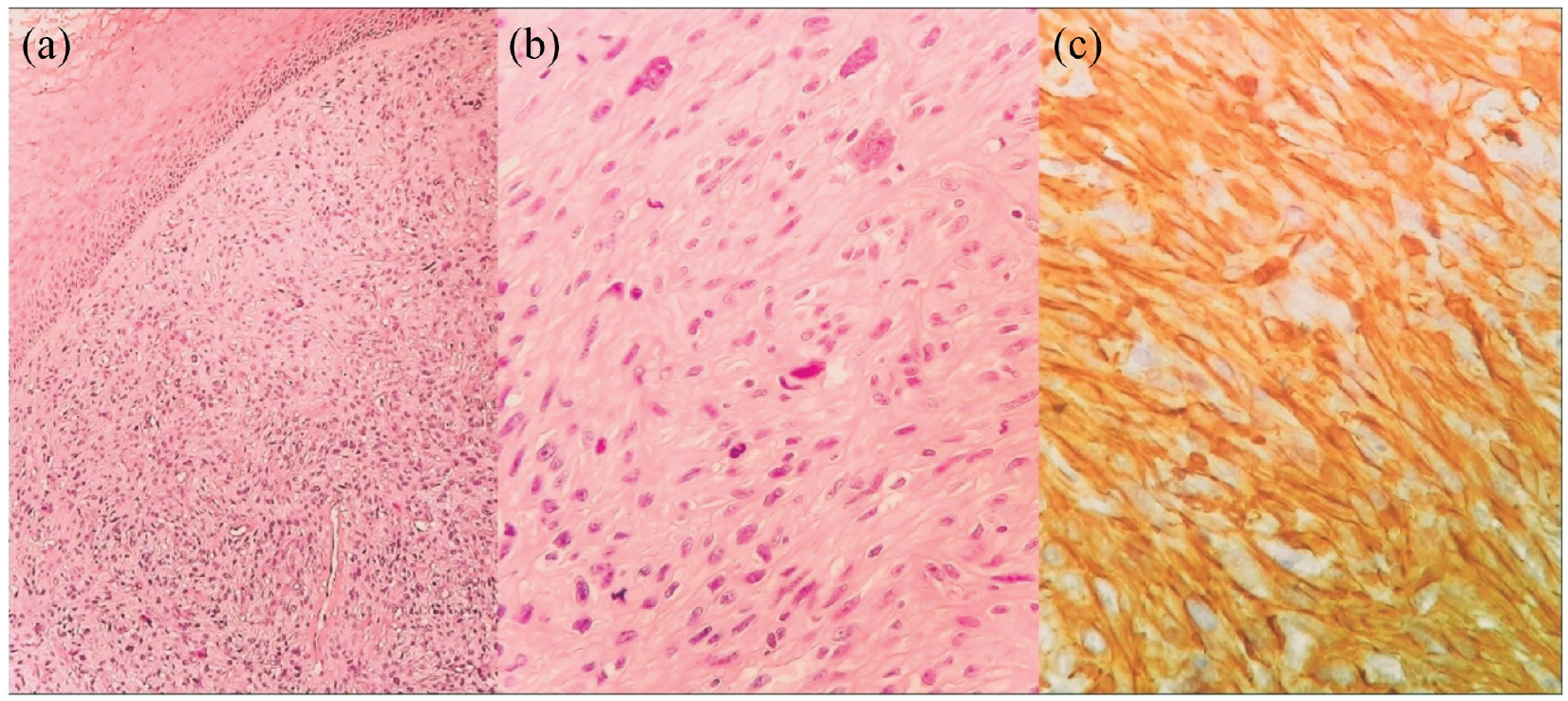
(a) Dermal spindle cell tumour. (b) Tumour cells exhibit marked nuclear atypia and numerous mitoses. (c) Tumour cells strongly express smooth muscle actin and h-caldesmon.
A thorough work-up was performed with no metastasis found.
The tumour was surgically removed with 1- cm safety margins and the subsequent skin defect was left for controlled wound healing.
The final pathological report confirmed the diagnosis of cLMS with tumour-free excision margins.
At the last follow-up, 1 year after surgery, the patient was doing well without any clinical signs of local or distant recurrence (Figure 3).

Clinical result at the last follow-up.
Discussion
Primary cLMSs are extremely rare, accounting for 2%–3% of all cutaneous soft tissue sarcomas.1–4 They are thought to be originating from normal arrector pili muscle. These tumours mainly affect men, with an average age of about 55 years.3,8,9,11,12 Cutaneous LMS occur on any part of the body mainly affecting the lower limbs (50%–75%), the upper limbs (20%–30%) and the trunk (10%–15%).10–14 The face is a rare location for cLMS and is only involved in approximately 1%–5% of the cases.9,10
A PubMed database search using the keywords ‘Cutaneous’ and ‘Leiomyosarcoma’ over the last decade (2013. . .) only finds 11 other case reports of cLMSs. To the best of our knowledge, our case is the 12th case overall and only the second located at the face (Table 1).
Recent cLMS case reports (10-year literature review).
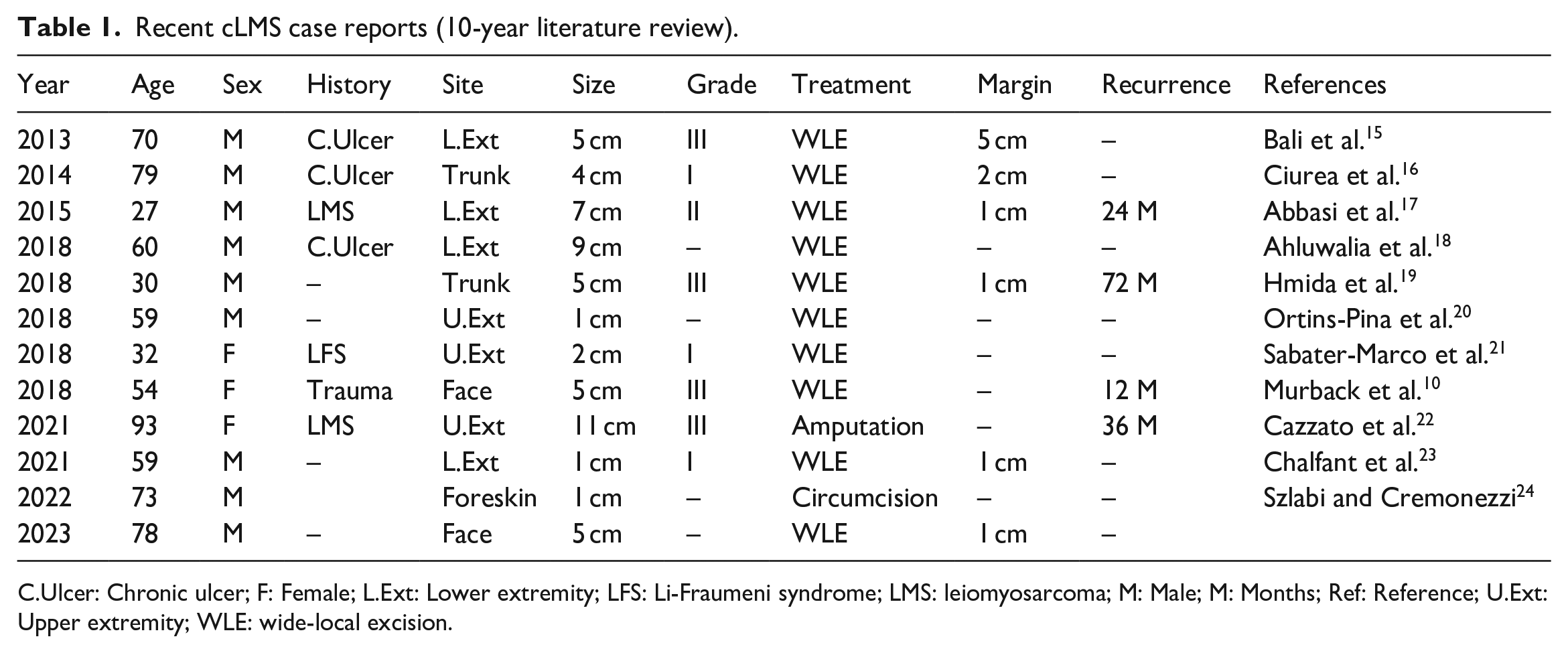
C.Ulcer: Chronic ulcer; F: Female; L.Ext: Lower extremity; LFS: Li-Fraumeni syndrome; LMS: leiomyosarcoma; M: Male; M: Months; Ref: Reference; U.Ext: Upper extremity; WLE: wide-local excision.
In most cases, the clinical diagnosis of cLMS is difficult due to the lack of specific features. In fact, this lesion can present either as a painless nodule or as a bleeding ulcerated and painful tumour, exceptionally as a bleeding ulcerated and proliferative bulging mass suggestive of a large pyogenic granuloma as is the case in our patient.25,26 Due to this variety of clinical presentations, the definitive diagnosis depends on histopathological findings.
Histologically, cLMS presents with malignant spindle-shaped cells, with eosinophilic cytoplasm and ‘cigar-shaped’ nuclei. Most are well to moderately differentiated. Mitoses are easily found and usually more than two mitoses per 10 high-power fields are identified, especially in the deeper tumours. 10 On immunohistochemical study, these tumours classically exhibit positive reaction with desmin, smooth muscle actin and H-caldesmone. Two histopathological growth patterns have been described by Kaddu et al.: either nodular or diffuse. 13
The aetiology of cLMS remains unknown, although predisposing factors including leiomyomas as precancerous lesions and history of previous injury or local trauma have been reported.12,23,27 Some authors have described primary cLMSs developing on scar tissue. 28
In general, the prognosis of LMSs is different whether they are cutaneous or subcutaneous. In fact, many studies suggest that dermal LMSs are more prone to local recurrence rather than distant metastasis and should, therefore, be reclassified as atypical smooth muscle tumours rather than sarcomas.8,13,29 However, Planet et al. 30 and Winchester et al. 9 reported a metastasis rate of 9% and 10%, respectively, with a mortality rate of 85% and 40%. Furthermore, Planet et al. in a multicentre, retrospective study of 79 patients treated for a cLMS over a period of 20 years found the following histological parameters to be associated, in univariate analysis, with a higher risk of local recurrence or metastasis: French Fédération Nationale de Lutte Contre le Cancer (FNCLCC) grades II or III (HR 4.8; 95% CI [1.6–14.6]; p = 0.01), a moderate or poor tumour differentiation (HR 4.5; 95% CI [1.2–17.2]; p = 0.03) and a high mitotic index (HR 4.0; 95% CI [1.3–12.5]; p = 0.02). 30
Cutaneous LMS metastasize more frequently to the lung and the skin,9,12 less commonly to bone, colon and lymph nodes. The mean survival at 5 years is about 92%. 3
As far as the treatment options are concerned, most authors agree that wide-local excision is the gold standard for cLMS. However, the optimal width and impact of the margin of resection on outcome are still subject to controversy. 31 A minimum 1-cm safety margin for resection is widely recommended; however, some authors consider that up to 3–5 cm are the required safety margins to minimize the risk of recurrence and metastasis.31,32 The in-depth excision should also be as complete as possible reaching up to the fascia and even removing the underlying muscle in the more infiltrating cases. Micrographic surgery, as described by Mohs, has also been reported as a valid therapeutic approach;8,9,25,26 however, recurrence rates of up to 14% have been seen with this technique.
Careful pathological examination of surgical margins is key in establishing the possibility of future recurrence. If the margins are not clearly negative, surgical re-excision is to be undertaken repeatedly if necessary. 32
There are no specific recommendations for the adjuvant therapy of cLMS, which remains non-standardized and largely anecdotical. 31 According to Kazlouskaya et al., indications for adjuvant treatment are high-grade tumours and recurrent or large tumours (>5 cm). 25 Other authors recommend radiotherapy in case of incomplete surgical resection with positive margins. As the occurrence of metastases from cLMS is a rare event, the best strategy for managing these patients is still subject to debate, and data on systemic treatments, such as chemotherapy, are scarce in the literature. 14 These rare cases of metastatic tumours should be managed with a multidisciplinary approach.
Conclusion
In general, cLMSs have a good prognosis, and surgical excision with at least a 1- cm safety margins allows complete recovery. However, some cLMS may metastasize, suggesting that wider local excision and long-term follow-up are necessary.
Footnotes
Acknowledgements
We would like to thank Dr Monia Tangour for the pathology figures as well as Mr Slim Jerbi for his help with English language processing.
Authors contribution statement
All the authors have equally contributed to the conception, drafting, writing and critical reviewing of the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Ethical approval for the study was granted from the Ethics Committee at the MT Maamouri Hospital, 130 Nabeul; TUNISIA (Reference: 75/2023).
Informed consent
Written informed consent was obtained from the patient for his anonymized information to be published in this article.