
Abstract
Nager syndrome (MIM #154400) is a rare acrofacial dysostosis syndrome predominantly characterized by malformations in craniofacial and preaxial limb bones. Most cases are sporadic and present with significant clinical heterogeneity. Although autosomal recessive and autosomal dominant modes of inheritance have been reported, most cases of Nager syndrome are spontaneous. Heterozygous variants in SF3B4 on chromosome 1q21 are found in approximately 60% of patients. Here, we report a first patient from Georgia diagnosed with Nager syndrome with detailed description of its clinical manifestations and diagnosis.
Introduction
Nager syndrome (MIM #154400, 201170) is an exceedingly rare genetic condition with around 100 reported cases in the medical literature. It is a part of disorders collectively termed dysostosis with predominant craniofacial involvement.1,2 The pattern of malformations in this condition is consistent with abnormal development of first and second branchial arches. The major facial features of Nager syndrome include downslanting palpebral fissures, malar hypoplasia, retrognathia and/or micrognathia, glossoptosis, left palate deformities, low-set, dysmorphic pinnae, microtia, external auditory canal stenosis/atresia and middle ear anomalies. Limb defects typically involve the anterior (radial) elements of the upper limbs and manifest as hypoplasia or absence of thumbs, triphalangeal thumbs, radial aplasia or hypoplasia and radioulnar synostosis.3–5 Intelligence is usually normal. Nager syndrome is caused by haploinsufficiency of SF3B4, encoding a spliceosomal protein SAP49. Many reported cases have been sporadic. A few families apparently with either autosomal dominant or recessive inheritance have been also described. 6 Here, we report the first case of Nager syndrome described in Georgia.
Case report
We describe a case of a 4-month-old girl who was referred to our hospital for clinical evaluation. The patient is the third child of a healthy non-consanguineous parents. She was born by C-section at 40 weeks of pregnancy. On 28th week of gestation prenatal ultrasound revealed an intrauterine growth retardation. The rest of the pregnancy was uncomplicated. Mother’s medical history revealed two medical abortions. Her history regarding alcohol, smoking, and drug abuse was negative. The patient was born with birth weight 2400 g and length 48 cm. Moreover, the child had feeding difficulties due to malformations of the orofacial region and kept in neonatal intensive care unit (NICU) for 21 days.
Clinical examination at the age of 4 months revealed a child with growth retardation evidenced by a height of 55 cm (3rd percentile), weight of 4100 g (below the 3rd percentile) and a head circumference of 47 cm (N). Cognitive development was normal. Craniofacial anomalies included microretrognathia, downslanting palpebral fissures, high forehead, low-set, posteriorly rotated and dysplastic cup shaped ears, absence of medial lower lid eyelashes, smooth philtrum, and high arched palate (Figure 1(a)–(c)). In addition, an absence of right thumb and right second finger duplication were observed (Figure 2(a) and (b)). Feet examination revealed inward rotation of the halluces (Figure 2(c) and (d)).
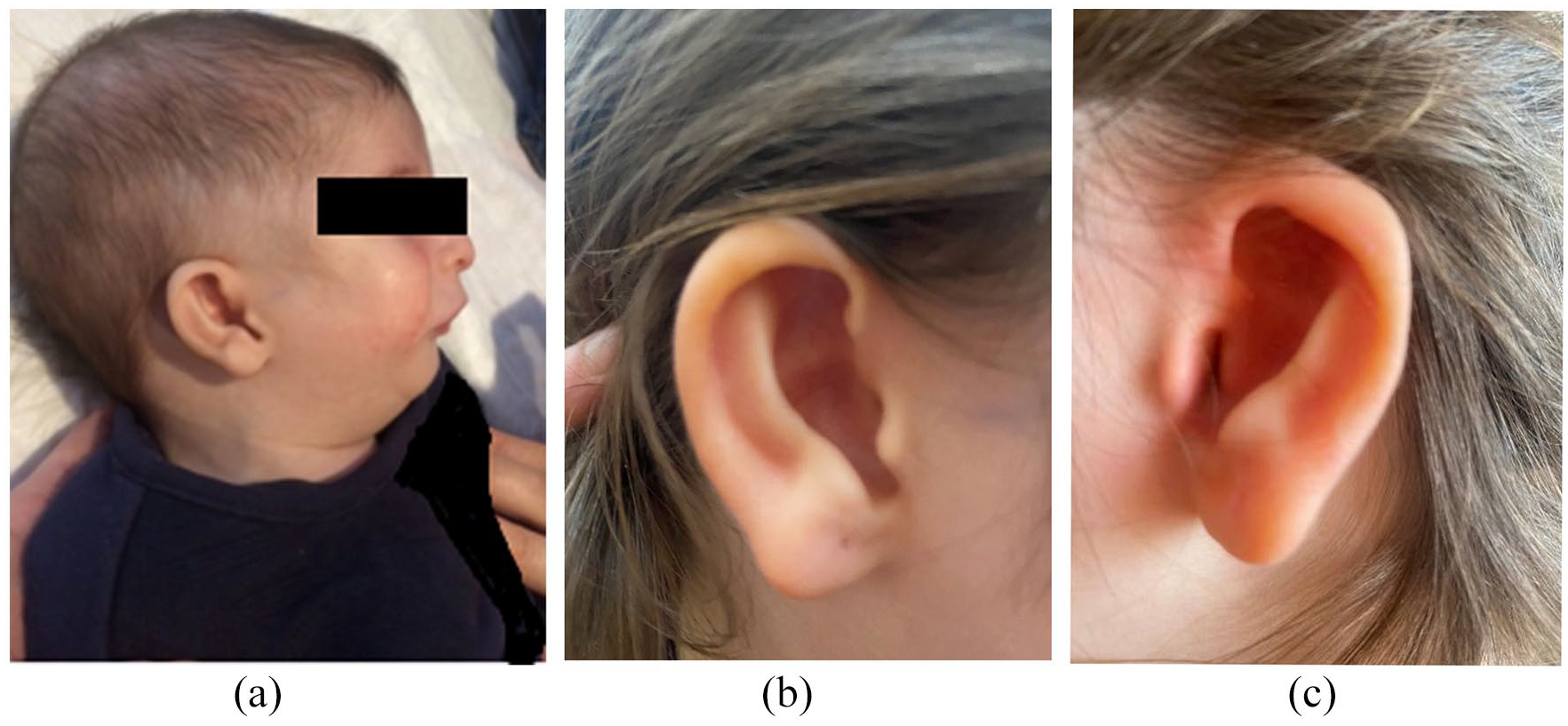
(a) Facial features of the patient showing retromicrognathia and low-set posteriorly rotated ears at 4 months. (b, c) Dysplastic and cup-shaped ears of the patients at 5 years.
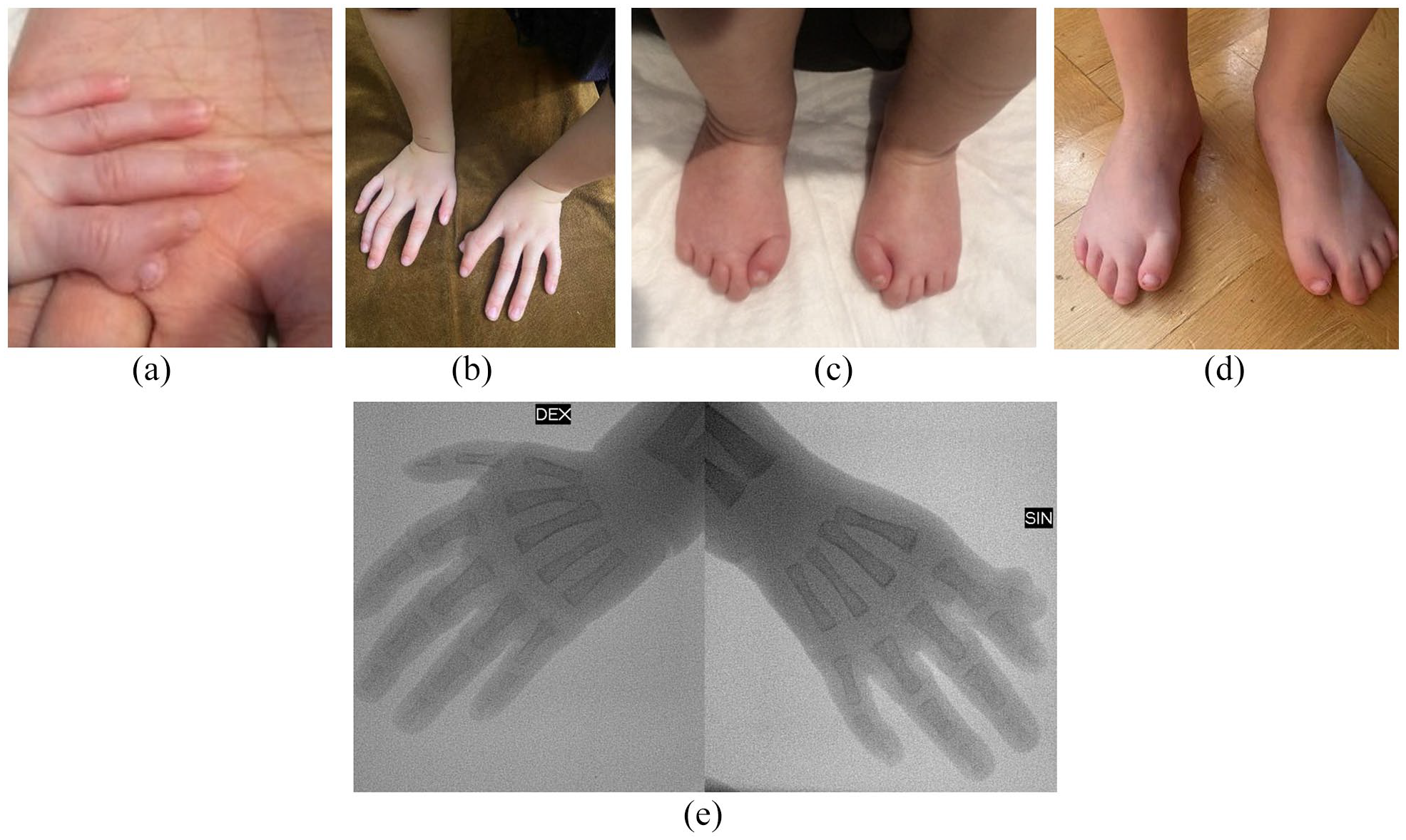
Hands of the patient showing absence of right thumb and right second finger duplication at 4 months (a) ad 5 years (b). Feet and toes showing inward rotation of the thumbs at 4 months (c) and short thumbs (d). X-ray of hands showing radial hypoplasia, absence of right thumb and duplication of right second finger (e).
Based on the patient’s craniofacial characteristics and the coexisting upper limb preaxial anomalies, a diagnosis of Nager syndrome was suspected. Routine hematological and biochemical tests were normal. Brain ultrasound, abdominal ultrasound and echocardiography were normal. Radiographic examination showed radial hypoplasia, absence of right thumb and duplication of right second finger (Figure 2(e)). Proximal radioulnar synostosis has not been seen. Malar and mandibular bones were hypoplastic. Cytogenetic analysis revealed a normal female karyotype – 46, . Whole exome sequencing (WES) was performed, which revealed a heterozygous de novo variant c.900del, p.(Met301Cysfs*19) in the SF3B4 gene. De novo status was confirmed by parental testing.
Upon clinical examination at the age of 5 years the patient exhibited hypernasal speech, nasal emission, and compensatory substitutions. Consonant and vowel distortion were present. Audiometric testing showed mild conductive hearing loss. A clinical oral examination revealed microdontia of the primary dentition. Serial ophthalmological examinations were normal. Currently, the patient is under the care of multidisciplinary team. Individualized speech/language therapy and intensive dental therapy and orthopedic management is planned.
Discussion
Facial dysostosis is a group of congenital craniofacial anomalies developing as a result of abnormal development of the first and second pharyngeal arches during embryogenesis. Nosology committee of the International Skeletal Dysplasia Society in its final revision of nosology and classification of genetic skeletal disorders classifies a group of disorders called dysostosis with predominant craniofacial involvement. 2 This group includes mandibulofacial dysostosis (Treacher Collins, Franceschetti–Klein); mandibulofacial dysostosis with microcephaly; mandibulofacial dysostosis with alopecia; Miller syndrome (postaxial acrofacial dysostosis); acrofacial dysostosis; Nager type; acrofacial dysostosis; Rodriguez type; acrofacial dysostosis; Cincinnati type; frontonasal dysplasia, types 1, 2 and 3; craniofrontonasal syndrome; acromelic frontonasal dysostosis; hemifacial microsomia; Richieri–Costa–Pereira syndrome; auriculocondylar syndrome, types 1, 2 and 3; orofaciodigital syndrome type I (OFD1) and Weyers acrofacial (acrodental) dysostosis. Facial dysostoses can be further classified into two types: mandibulofacial dysostoses (MFDs) and acrofacial dysostoses (AFDs). MFDs typically have no limb abnormalities, while AFDs are present with limb defects. Treacher Collins syndrome (TCS) is a very well-characterized disorder of craniofacial development belonging to MFDs that leads to a similar facial phenotype as Nager syndrome which is a best example of AFDs. The Nager syndrome was first described as distinct from TCS by Nager and De Reynier in 1948 and manifests from lesions occurring between weeks 3 and 4 of blastogenesis. Its major clinical manifestations are:1,7,8
Craniofacial dysmorphism: malar hypoplasia, downslanting of the palpebral fissures, lower eyelid coloboma, micrognathia, ptosis of upper lids, deficiency of eyelashes of the medial one- to two-thirds of the lower eyelids, cleft or high arched palate, ear malformations such as low- set, posteriorly rotated, dysplastic ears, preauricular tags and external auditory canal atresia.
Upper limb anomalies: hypoplasia or absence of thumbs, triphalangeal thumbs, radial aplasia or hypoplasia, radioulnar synostosis, short forearms, very rarely phocomelia.
Conductive hearing loss attributed to malformations of the auricle and external auditory canal. Occasionally mixed hearing loss.
Mostly normal lower limbs, but minor foot deformities may be present, including metatarsus varus, short halluces, missing, hypoplastic, overlapping toes, syndactyly, broad hallux, clubfeet; fibular aplasia in lethal cases.
It has been confirmed that haploinsufficiency of SF3B4 is an accepted disease-causing mechanism for Nager syndrome.9,10 Haploinsufficiency is defined as a dominant phenotype in diploid organisms that are heterozygous for a loss-of-function (LOF) allele. Similar mechanism has been described for another mandibulofacial dysostosis caused by EFTUD2 gene, encoding spliceosomal GTPase. 11 The SF3B4 gene encodes one of subunits of the splicing factor 3b (SF3b). SF3b consists of seven subunits: SF3b1, SF3b2, SF3b3, SF3b4, SF3b5, SF3b14, and PHF5A, among which the SF3b4 gene (1q12-q21), encodes spliceosome-associated protein 49 (SAP49), which is the core subunit of U2 snRNP and plays an important role in the pre-mRNA splicing by stable binding the U2 snRNP to the branchpoint sequence (BPS) in pre-mRNA. 12 Approximately 60% of patients with features of Nager syndrome have pathogenic variants in SF3B4 gene, which are mostly frameshift, nonsense and splice site mutations.9,10,12,13 For the rest of the patients, the cause remains unknown suggesting existence of genetic heterogeneity. Most cases represent sporadic events, likely due to a de novo mutations, although both autosomal dominant and autosomal recessive inheritance have also been described.14,15
Interestingly, the c.900del, p.(Met301Cysfs*19) variant detected in our patient in located in exon 4 of SF3B4 gene and creates a shift in the reading frame starting at codon 301. The new reading frame ends in a stop codon 18 positions downstream, thus it causes premature termination of the translation and consequently is considered to be LOF, consistent with haploinsufficiency mechanism. This variant is absent in the databases like genomAD, a large reference population database which aims to exclude individuals with severe pediatric disease, and in 1000G. The variant did not segregate in any of the healthy parent, confirming de novo nature. The above-mentioned characteristics supporting pathogenicity of the detected variant.
Diagnosis of Nager syndrome relies on clinical features, patient history, imaging studies and genetic testing. Distinguishing it from other mandibulofacial disorders may be challenging based on phenotype alone. Thus, broad molecular-genetic investigations, like WES, may prove to be a valuable tool for establishing the correct diagnosis.
Patients with Nager syndrome require a complex management and should be tailored to the specific needs of each patient. Early diagnosis can improve the quality of life. A multidisciplinary team involving neonatology, pediatrics, oral, hand and plastic surgery, pediatric otolaryngology, otology, audiology, psychology, and genetics is best suited to care for this challenging disorder.
Conclusion
Nager syndrome is a rare genetic condition with craniofacial and preaxial limb involvement. Most cases appear to be sporadic, although both autosomal dominant or autosomal recessive inheritance have been documented. Distinguishing Nager syndrome from other mandibulofacial disorders can be difficult based on phenotype alone therefore extended genetic testing like whole exome sequencing (WES) is an important to exclude other diseases with similar clinical features. Timely and correct diagnosis is important for proper medical intervention, coordinated multidisciplinary care, anticipatory guidance and genetic counseling, resulting in improved quality of life for patients with Nager syndrome and their families.
Footnotes
Acknowledgements
The authors thank the patient and parent for granting permission to publish this information.
Author contributions
T.T., K.B. and E.A. conceptualized the report. E.K., M.A.K.and N.O. wrote the manuscript. All authors have read and approved the submitted version of this manuscript.
Data availability
The data used in this paper is available from the corresponding author (EK) on request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the legal representative of the patient for the anonymized information to be published in this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.