
Abstract
A synchronous tumor represents two histologically distinct neoplasms occurring at the same anatomic site, each displaying a distinct tumorigenesis pathway; they can be primary, secondary, or a mixture. The occurrence of an adenocarcinoma and lymphoma has been reported in gastrointestinal and pulmonary sites; however, such a finding in hepatobiliary system remains elusive. Primary hepatic lymphoma is rare, while primary biliary mucosa-associated lymphoid tissue lymphoma is an even rarer event; hence, its collision with an intrahepatic cholangiocarcinoma could be entirely missed both in practice and in the literature. We herein reported a case of biliary mucosa-associated lymphoid tissue lymphoma occurring synchronously with an intrahepatic cholangiocarcinoma in a 78-year-old female following a biopsy-proven intrahepatic cholangiocarcinoma and hepatectomy. Microscopic examination identified atypical lymphoid population intermingled with this intrahepatic cholangiocarcinoma. An immunohistochemical panel uncovered an incidental mucosa-associated lymphoid tissue lymphoma occurring with this intrahepatic cholangiocarcinoma. This clinically missed entity led to comprehensive systemic investigation/staging, with subsequent detection of bone marrow involvement by stage IV lymphoma. This unique case highlights the importance of astute histomorphological evaluation and thorough ancillary studies in identifying a clinically unsuspected neoplasm in close contact with a known tumor.
Keywords
Introduction
A synchronous tumor is an extremely rare phenomenon that consists of two histologically distinct neoplasms occurring within the same organ with no significant overlap. This event phenomenon is unique in that both tumors usually display a different histogenesis and tumorigenesis pathway.1,2 A synchronous tumor frequently presents a diagnostic challenge pathologically and clinically. It should be distinguished from the tumors from a pluripotent stem cell that could demonstrate more than one lineage-specific tumor. 3 A thorough workup including a detailed histological examination with appropriate ancillary studies might be required to evaluate the differentiation of histologically different components within a single tumor. 4 Intrahepatic cholangiocarcinoma (IHCC) is an intrahepatic malignancy with biliary epithelial differentiation that can arise in any portion of the intrahepatic biliary tree, commonly in a non-cirrhotic background liver. IHCC is the second most common primary hepatic malignancy after hepatocellular carcinoma and accounts for approximately 5%–10% of primary liver cancers. 5
Primary hepatic lymphoma accounts for 0.016% of all cases of non-Hodgkin lymphoma, with diffuse large B-cell lymphoma (DLBCL) being the commonest subtype. 6 The majority of primary hepatic lymphomas are diagnosed as DLBCLs. Primary hepatic mucosa-associated lymphoid tissue (MALT) lymphoma is infrequently reported, approximately 70 cases documented per literature.7–10 It represents a unique subset of primary liver lymphoma with variable clinical presentation. 11 Similar to nodal marginal zone lymphoma and splenic marginal zone lymphoma, primary hepatic MALT lymphoma also consists of a heterogeneous group of neoplasms that resemble the normal B-cell populations of the marginal zone and frequently show monocytoid or plasmacytic differentiation and associated with chronic inflammatory processes12–14 Biliary cirrhosis, hepatitis C or B, and bacterial infection, for example, Helicobacter pylori, are considered to be the etiologies for primary hepatic MALT lymphoma.8,15,16 A case report highlighted an association with non-alcoholic steatohepatitis (NASH). 10 It is like that there is a relationship between autoimmunity and hepatic MALT lymphoma; however, there is no definite evidence to establish a causal relationship.
Case presentation
A 78-year-old female with a past medical history of diverticulitis and irritable bowel syndrome presented to an outside hospital emergency room due to abdominal pain. Initial workup revealed a liver mass on computerized tomography (CT) of the abdomen and pelvis. Additional abdominal magnetic resonance imaging (MRI) without contrast confirmed the presence of a 5.1 × 2.8 × 2.3 cm T2 hyperintense and T1 hypointense irregular mass in the lateral segment of the left hepatic lobe, which demonstrated increased signal on the diffusion weighted scan, raising the concern for an infiltrative intrahepatic neoplasm (Figure 1(a) and (b)). A separate CT scanning reveals an ill-defined low-density mass at the same location (Figure 1(c)). A CT-guided needle core biopsy of the liver revealed a tumor with closely packed small tubular and acinar structures in a desmoplastic stroma with a prominent inflammatory aggregate (Figure 2(a) and (b)). The tumor was positive for CK7, S100P (Figure 2(c) and (d)), and negative for HepPar 1 (Figure 2(e)), TTF-1, GATA3, PAX 8, CDX2, and Villin by immunohistochemical (IHC) study (images not shown), with a Ki67 proliferation index at 11.5% by manual morphometric quantitative analysis (Figure 2(f)). The morphological and immunohistochemical findings are consistent with adenocarcinoma, moderately to poorly differentiated, with pancreaticobiliary features and negative for synaptophysin, chromogranin, and P40, arguing against neuroendocrine neoplasm and squamous cell carcinoma.
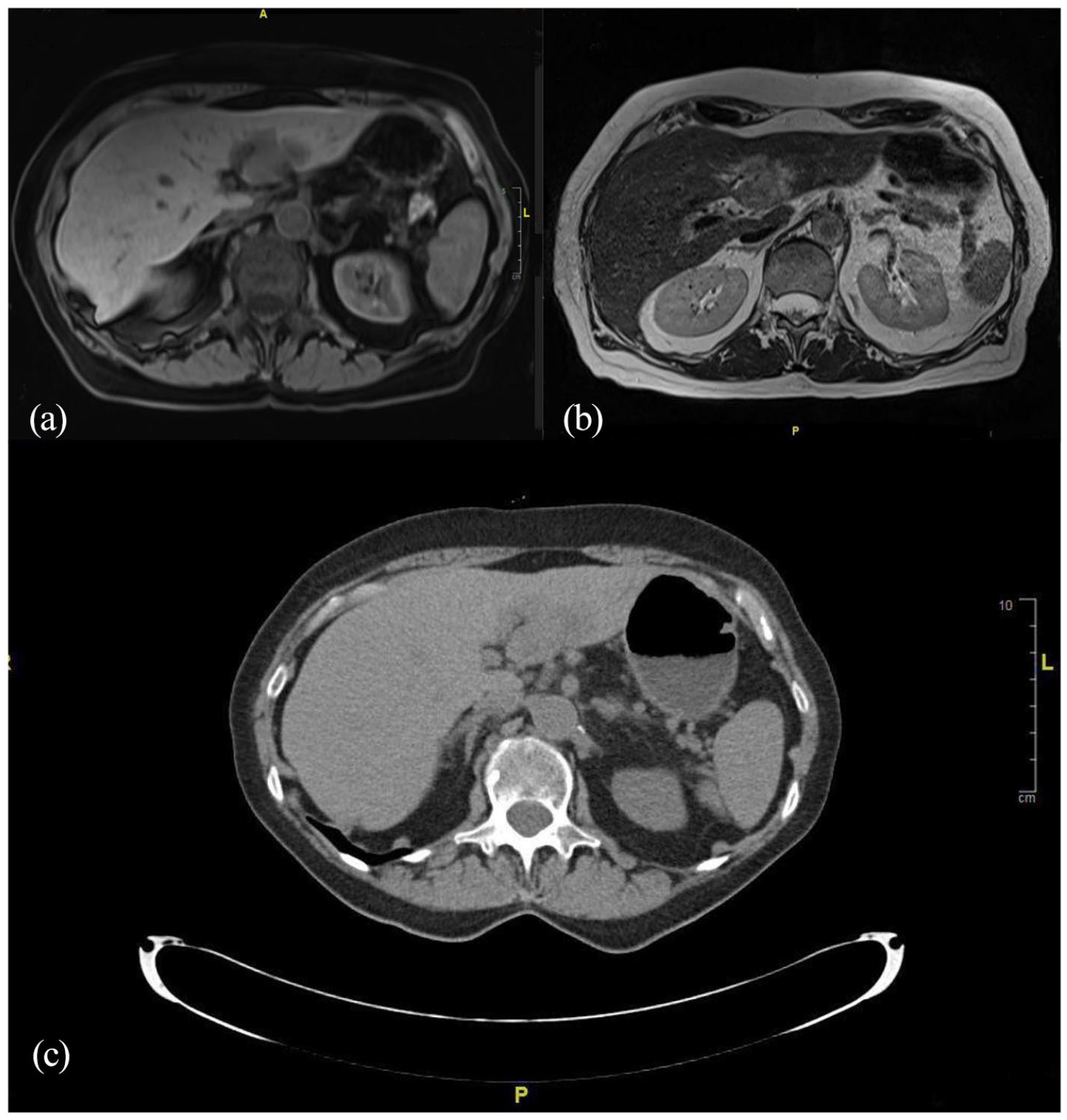
(a) Computerized tomography (CT) of the abdomen and pelvis revealed a left liver mass in a 78-year-old female. (b) Abdominal magnetic resonance imaging (MRI) without contrast confirmed the presence of a 5.1 × 2.8 × 2.3 cm T2 hyperintense and T1 hypointense irregular lesion in the lateral portion of the left liver lobe, which demonstrated enhanced signal on the diffusion weighted scan indicating an infiltrative intrahepatic neoplasm. (c) A follow-up CT scanning highlighted an ill-defined low-density mass at the same location.
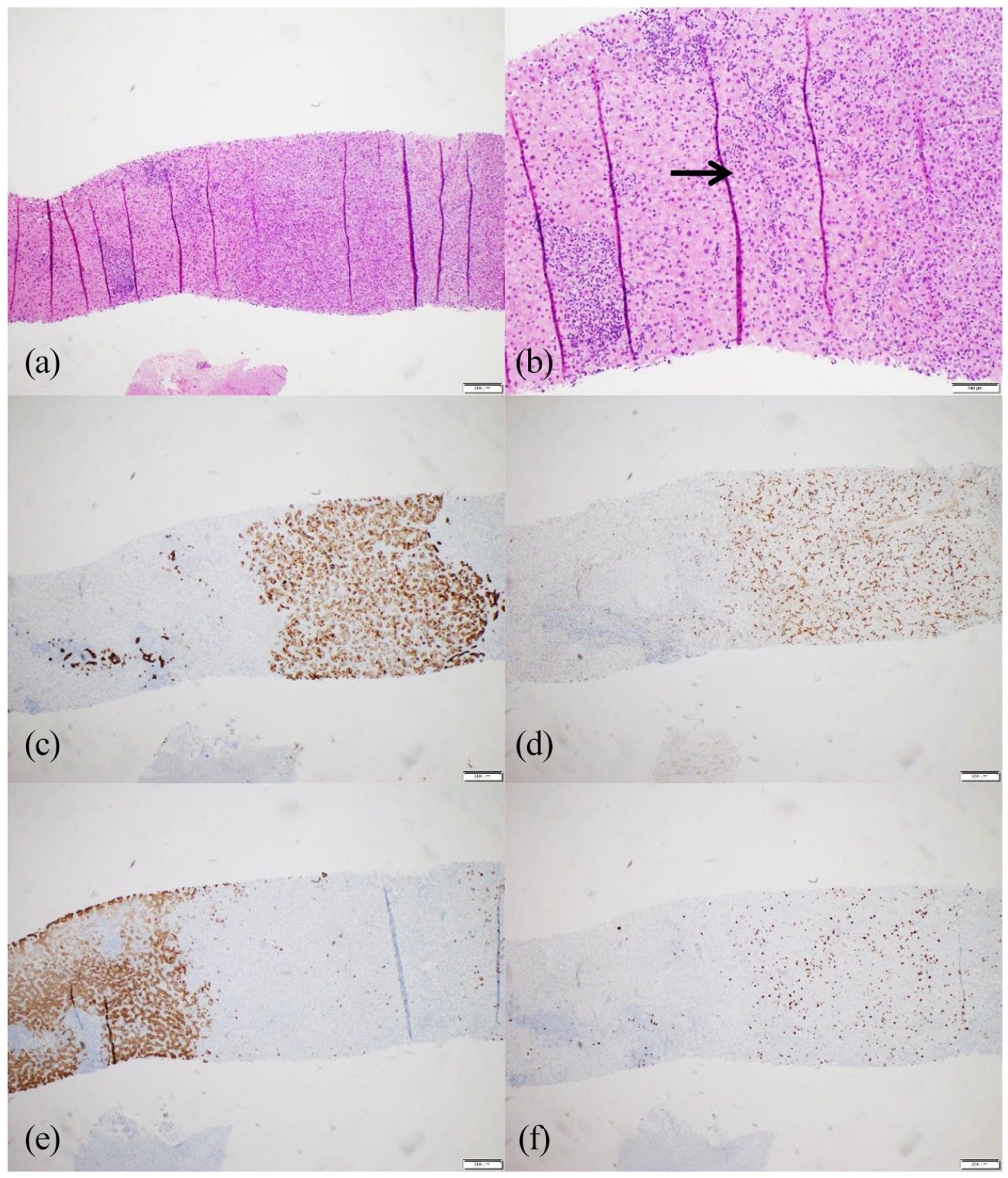
(a, b) A CT-guided needle core biopsy of the liver mass revealed a tumor composed of closely packed tubular and acinar structures in a desmoplastic stroma. Tumor cells were positive for (c, d) CK7, S100P, and negative for (e) HepPar 1, TTF-1, GATA3, PAX 8, CDX2, and Villin by immunostaining (images not shown), with a (f) Ki67 proliferation index at 11.5% by manual morphometric quantitative analysis. The morphological and immunohistochemical findings are most consistent with intrahepatic cholangiocarcinoma (IHCC).
The patient was referred to our institution for further management. Repeat CT of the thorax, abdomen, and pelvis performed at our institution confirmed the presence of the liver mass with features similar to those stated in the histopathology report from the referring hospital and no evidence of metastatic disease. The patient underwent exploratory laparotomy with left hepatectomy and portal and left gastric lymphadenectomy. Gross examination of resected liver revealed a pale tan, poorly circumscribed, intraparenchymal mass measuring 4.8 × 4.0 × 3.1 cm with a corresponding irregularly fibrotic area on the capsular surface overlying the tumor. Grossly the mass was 2.0 cm from the closest parenchymal resection margin and the remaining liver parenchyma was grossly unremarkable (image not included). Microscopically, this IHCC was composed of neoplastic small tubular glands, duct-like structures, and frequent areas of solid growth patterns embedded within a desmoplastic stroma (Figure 3(a) and (b)). The tumor is composed of low cuboidal to columnar cells with eosinophilic cytoplasm and round to oval nuclei (Figure 3(c)). Lymphovascular and perineural invasion were not identified after extensive sectioning. By IHC study, this IHCC was negative for HepPar1, arginase, CDX2, synaptophysin and chromogranin, and positive for pan-keratin (IHC images not shown); there were five benign lymph nodes with preserved nodal architecture. Tumor also showed intact nuclear staining for MLH1, MSH2, MSH6, and PMS2—this finding does not support microsatellite instability. Based on observed parameters, the tumor was assessed to be pT1aN0.

(a, b) This intrahepatic cholangiocarcinoma shows small glands and ducts embedded in solid growth patterns in close contact with markedly dense mononuclear infiltrate, (c–f) composed of monomorphous, medium-sized lymphoid proliferation. (d) These lymphoid populations show slightly irregular nuclei, moderately dispersed chromatin, inconspicuous nucleoli, and a moderate amount of pale cytoplasm. A few lymphoepithelial lesions were easily identified (arrows in d, e).
A dense monomorphous population of mildly atypical, medium-sized lymphoid cells was seen intimately admixed with the carcinomatous glands and surrounding stroma (Figure 3(c)–(f)). These lymphoid cells are small and show oval to slightly irregular nuclei, moderately dispersed chromatin, inconspicuous nucleoli, and a moderate amount of pale cytoplasm. A few lymphoepithelial lesions were identified (Figure 3(d) and (e)). By IHC study, these atypical lymphoid cells are positive for CD20 and BCL2 (Figure 4(a) and (b)), with a Ki67 proliferation index approximately at 5%–10% (not shown). These lymphoid cells are negative for CD3 and BCL6 (Figure 4(c) and (d)), CD43 and Cyclin D1 (Figure 4(e) and (f)); they are also negative for CD5, CD23, and LEF1 (these three markers are commonly positive in small lymphocytic lymphoma) (not shown). The histomorphological and immunophenotypical findings are characteristic for extranodal marginal zone lymphoma of MALT. Taken together, a diagnosis of synchronous tumor composed of IHCC and primary hepatic MALT lymphoma was finally rendered; the findings were swiftly conveyed to and discussed with the clinical team. Subsequent clinical workup revealed no hepatosplenomegaly, generalized lymphadenopathy, or extranodal masses, but this patient’s bone marrow was involved by a small population of neoplastic lymphocytes with identical histomorphological and immunophenotypical phenotypes to the MALT lymphoma, confirming a stage IV lymphoma clinically.

(a, b) By immunostaining these atypical lymphoid lesions are positive for CD20 and BCL2, with a Ki67 proliferation index at approximately 10% (not shown). They are negative for (c, d) CD3 and BCL6, (e, f) CD43 and Cyclin D1.
Discussion
IHCC originated from the small bile ducts within the liver is the second most common liver malignancy that commonly presents in the sixth decade of life with an increased prevalence in men.17,18 This cancer arises from the intrahepatic biliary tree, frequently in a non-cirrhotic background liver.17,19 Risk factors reported in the literature for development include liver fluke infection (Clonorchis sinensis and Opisthorchis viverrini), stones in the liver and biliary tract (hepatolithiasis), primary sclerosing cholangitis (PSC), and congenital bile duct anomalies17,18 Other risk factors also include a long-standing hepatitis B or C infection, diabetes, and obesity.20,21 It is debatable whether smoking and alcohol use are contributory. 21 Our patient’s past medical history includes diverticulitis and irritable bowel syndrome, with previous unremarkable colonoscopies. The patient did not have hepatitis B or C infection, PSC, and diabetes based on available clinical information. This patient’s former smoking history, approximately 20 years of 0.75 packs per day, presumably contributed to an increased risk for developing IHCC.
Extranodal marginal zone lymphoma of MALT is the most common type of marginal zone lymphoma accounting for 5%–8% of all B-cell lymphomas and has been described in many organs that usually are devoid of germinal centers; one possible etiology is that it arises from lymphoid populations that are induced by chronic inflammation in extranodal sites. 22 The chronic inflammation may be the result of infection, autoimmunity, or other unknown stimuli; the link between gastric MALT lymphoma and H. pylori infection has been well established.22,23 Chlamydia psittaci and Borrelia burgdorferi have been proposed as causative agents for ocular and cutaneous marginal zone lymphomas through their role in chronic antigenic stimulation. 22 Autoimmune-associated chronic inflammation in Sjogren’s diseases and Hashimoto’s thyroiditis is another known preceding event in salivary and thyroid gland MALT lymphoma, respectively. 24 It has been established that stomach is the most common site (35%) for MALT lymphoma, followed by the eyes and adnexa (13%), skin (9%), lung (9%), salivary gland (8%), breast (3%), and thyroid (2%). 12 A characteristic molecular finding in a subset of MALT lymphoma is frequent translocation of genes involving MALT1 and BCL10, which leads to the activation of the NF-κB signaling pathway. 25
Primary hepatic lymphoma is a very rare malignancy; although the liver contains lymphoid tissue, host factors make the liver an unsuitable environment for the development of lymphoma. 26 A causative role of HIV, hepatitis B, hepatitis C, Epstein–Barr virus, liver cirrhosis, primary biliary cirrhosis, immunosuppressive therapy, and autoimmune disease have all been implicated; however, pathogenesis of primary hepatic lymphoma remains poorly defined. 27 Based on the limited reports available, primary hepatic MALT lymphoma is extremely rare. 9 The aforementioned etiology, for example, hepatitis B or C or H. pylori, for primary hepatic MALT lymphoma is not found in our case. Instead, sustained chronic inflammatory processes, for example, diverticulitis and irritable bowel disease, were identified, which may be the cause for an abnormal B-cell proliferation in the hepatobiliary system eventually leading to MALT lymphoma. Nonetheless, it is assumed that the inflammation is also associated with IHCC, suggesting the synchronous tumors may share the same etiology as inflammatory or immune stimulation is now recognized as an important parameter of lymphomagenesis. 14
Of note, the initial biopsy of the liver mass in our case performed at an outside institution did not reveal an MALT lymphoma, likely due to small sample size, limited material, and tissue crushing artifact. A high-level index of suspicion, when presented with monotonous proliferation of small or plasmacytoid lymphocytes in an unusual site such as liver and in conjunction with another malignancy, is pivotal to diagnose MALT lymphoma. A detailed clinical history and thorough workup including flow cytometry and immunohistochemical studies are crucial to establish an accurate diagnosis and to provide proper management. Treatment of MALT lymphoma depends on the stage of disease. Stage I and II gastric MALT lymphoma are initially treated with H. pylori eradication. 22 Similarly, ocular MALT lymphoma shows regression in approximately 65% of cases with doxycycline. 22 Patients with disseminated stages III and IV disease with systemic symptoms are treated with chemotherapy (e.g. bendamustine, fludarabine, or chlorambucil) combined with either an anti-CD20 antibody rituximab or immunomodulatory drug lenalidomide. 25 There are no established therapeutic protocols or guidelines for the treatment of primary hepatic MALT lymphoma. Surgery, chemotherapy, or radiotherapy alone or in combination have been described in the literature. 8 Since bone marrow biopsy in our case was positive for involvement by low-grade B-cell lymphoma, it usually warrants administration of systemic chemotherapy with anti-CD20 antibody rituximab. Complete surgical resection is the sole potentially curative treatment option for managing IHCC. 28 Our patient underwent surgical resection with negative surgical resection margins, and no lymphovascular involvement was identified. Treatment with 6 months of adjuvant capecitabine was recommended, which has been shown to have an increased overall survival in the literature. 29 There is no recurrent disease after 11 months of follow-up for our patient.
Conclusion
This novel IHCC colliding with liver MALT lymphoma case represents an interesting finding that pathologists and clinicians should be aware of when managing patients with an IHCC intermixed with prominent inflammatory infiltrate. Astute histomorphological examination combined with essential ancillary investigations are paramount in reaching a complete and accurate classification for optimal patient management.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.