
Abstract
Intravenous immunoglobulin is a recognized treatment in recalcitrant autoimmune bullous diseases. Infusions are administered monthly over 1–5 days in the hospital setting and associated with mild to severe infusion-related systemic effects, in part due to the high doses necessary to induce and achieve remission. We present a case series of four patients with bullous diseases treated successfully with low-dose subcutaneous IgG who achieved remission with maintenance therapy. Patient-administered smaller, more frequent doses of IgG into subcutaneous tissue more closely mimics the body’s own antibody production and produces a more stable serum trough level. Subcutaneous IgG is a novel treatment approach in bullous diseases which can induce a state remission.
Introduction
Autoimmune bullous diseases are characterized by bullous lesions to the skin and mucous membranes of the oral cavity, nose, eyes, larynx, pharynx, esophagus, and genitals. 1 Once considered fatal conditions, treatment options have evolved from corticosteroids to steroid-sparing immunosuppressant drugs (methotrexate (MTX), azathioprine and mycophenolic acid (MMF)), monoclonal antibodies (rituximab), and intravenous immunoglobulin (IVIG).2–4
IVIG is a human plasma derivative containing IgG and has been used in conjunction with conventional therapy to treat refractory bullous diseases.2,4 Subcutaneous IgG (SCIG) is an effective alternative for patients refractory to or unable to tolerate immunosuppressive therapy. 4 Moreover, it has been shown to be more cost-effective than immunosuppressives, which can result in significant toxicities requiring hospitalization. 5 The immunomodulatory effects are complex and multifaceted, including increased catabolism of autoantibodies, inhibition in autoantibody function, and decrease in plasma inflammatory markers.2,3,6 Optimal dosing varies but follows similar conventional weight-based approaches (300–400 mg/kg/month) as well as higher doses (2 g/kg over 2–5 days/month) in aggressive disease.2,4,7 Adverse events can be mild (headaches, backaches, hives), severe (anaphylaxis, thromboembolism), and are usually infusion-related and self-limiting. However, adverse events increase with higher doses and may interfere with patient’s quality of life.3,6
The goal of therapy in the bullous diseases is to induce and maintain remission, as evidenced by the cessation of new vesicle and bullae formation and healing of old lesions.3,8 Long-term therapy may be required in recalcitrant disease and may be associated with significant toxicities if corticosteroids or immunosuppressants are needed, particularly in elderly patients with bullous pemphigoid. 8 In the present case series, we describe the use of low-dose SCIG (Hizentra; CSL Behring Inc) to safely induce and maintain long-term remissions in four patients with biopsy and immunofluorescence confirmed autoimmune bullous diseases. All diagnoses were confirmed by a dermatologist and pathology.
Case series
Case 1
A 58-year-old woman with 15 years history of linear IgA disease presenting with bullous lesions (oral, nasal, ocular, and vulvar mucosa), severe burning, pruritis, and pain to the affected areas necessitating the utilization of dark glasses because of photosensitivity. Initial treatment with dapsone led to a hemolytic anemia and hospitalization secondary to G6P dehydrogenase deficiency. Prednisone, sulfapyridine, and IVIG, 125 g IV (1 g/kg) over 2 days monthly, were effective but required time from work to accommodate IVIG infusions and manage the severe side effects (nausea and headaches). Her disease would flare 2–3 weeks post monthly IVIG, later acquiescing with every 2-week treatment (55 g). Eventually, prednisone was stopped, and IVIG further reduced (25 g every 2 weeks). She found IVIG inconvenient and transitioned to self-administered SCIG 8 g weekly (tapered to 8 g every 10 days after 2 months (24 g/month)). As shown in Figure 1, as compared to IVIG, plasma IgG levels remained stable with low-dose SCIG with no side effects and excellent disease control. After 3 years on SCIG, her sulfapyridine was stopped. She is working full time and has undergone gastric bypass surgery with a subsequent 50 kg weight loss.

Patient’s IgG trend over time.
Case 2
A 63-year-old woman referred with bullous pemphigoid, refractory to prednisone (50 mg) and oral MTX with bullous lesions to her legs, torso and perineum, with intractable pruritis (Figure 2(a)). There was widespread scarring to affected areas from scratching and she was unable to return to work. She was treated with multiple courses of rituximab (375 mg/m2) and 50 g IVIG monthly (1 g/kg) and experienced severe pruritis and urticaria requiring antihistamines and analgesia. While on rituximab every 3 months and prednisone 15 mg/day, her abdominal blisters recurred and SCIG (3 g/week) was started. She achieved complete remission (Figure 2(b)) for the subsequent 18 months, allowing discontinuation of rituximab and prednisone.
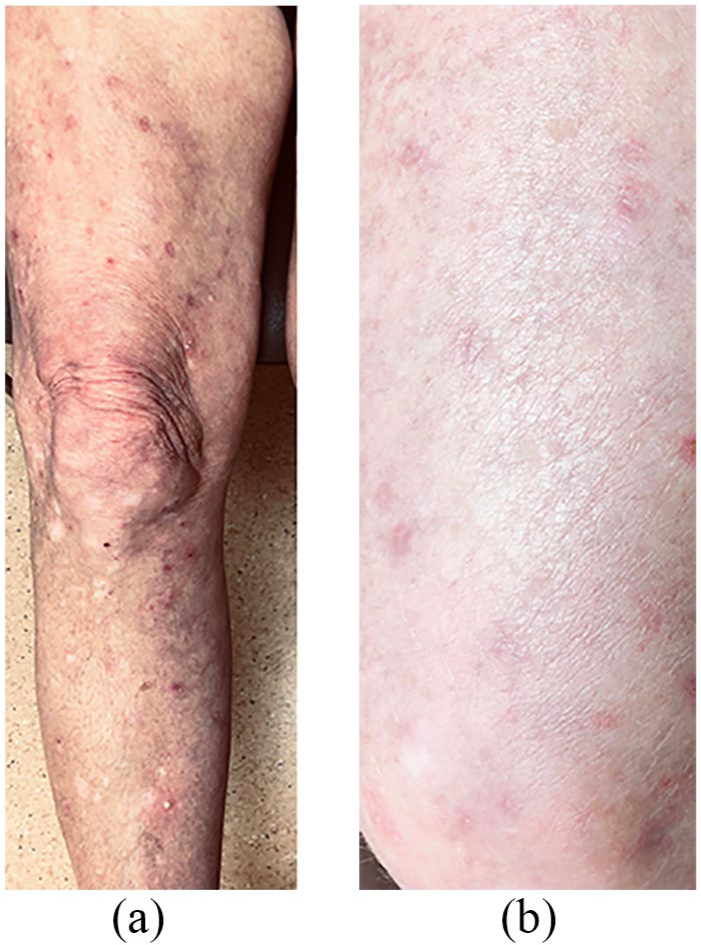
(a) Pre-SCIG and (b) post-SCIG.
Case 3
An 85-year-old woman with 9-year history of bullous pemphigoid refractory to MMF, azathioprine, and dexamethasone swish and spit mouth rinses referred with ulcerations on her buccal mucosa, soft palate and arms. She started SCIG 4 g/week and continued azathioprine 50 mg orally twice daily. Three months later, her ulcerations resolved, and no further bullous skin lesions developed and her azathioprine was tapered to 50 mg/day. Two months later, azathioprine was stopped and SCIG dose tapered to 3 g/week. The patient has been on SCIG for 12 months.
Case 4
A 63-year-old man with pemphigus vulgaris for 3 years had progressive disease with oral mucosal and chest ulcerations despite weekly rituximab, MTX (2.5 mg/day), and prednisone 25 mg/day. Despite efficacy of a 6-month course of IVIG 90 g (1 g/kg) and prednisone 20 mg/day, the patient experienced significant back pains and headaches requiring pre-infusion steroids. Switching to SCIG twice weekly (5 g infusions) led to disease control and excellent tolerance. Prednisone was tapered after 4 months and discontinued at 1 year. He remains in complete remission on 6 g/week SCIG.
Discussion
In this case series, we describe four patients with autoimmune bullous diseases, who achieved complete, sustained remissions with low-dose SCIG allowing other immunosuppressants to be discontinued or reduced. Recommended IVIG dosing is 2 g/kg/month, while we found that 12–40 g/month SCIG (which maximizes volume/site) was effective and could be further reduced over time. 2 Intravenous delivery produces an initial spike in plasma IgG levels, equilibrating between the intra- and extra-vascular circulations in 3–5 days. In contrast, SCIG is gradually absorbed over 3–5 days producing a sustained plasma IgG level (Figure 1).9,10 Thus, smaller more frequent dosing and slower absorption results in more stable serum IgG trough levels with SCIG, potentially explaining the lack of side effects and pre-medications which are associated with high peak levels, but retained clinical benefit. 11
SCIG has been widely adopted for primary and secondary hypogammaglobulinemias.10–13 We demonstrate that administering SCIG at home has proven advantages (Table 1).14–16 The strengths of this case series are that to our knowledge this is the first report demonstrating the effectiveness of SCIG as adjuvant or maintenance therapy in the treatment of recalcitrant bullous conditions. Moreover, we utilize much lower doses than with IVIG while demonstrating fewer adverse effects and increased savings. While findings from a small case series cannot be generalized without a randomized control trial or clinical trial, we are pursuing further inquiry with SCIG as a novel systemic treatment option in this patient population.
Advantages of SCIG.

SCIG: subcutaneous IgG; IV: intravenous; IgG: immunoglobulin G.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Informed written consent was obtained from all individuals included in this case series.