
Abstract
Primary angiitis of the central nervous system is a rare idiopathic vasculitis affecting small- and medium-sized vessels, isolated to the brain, leptomeninges and spinal cord. We report a case of biopsy-proven primary angiitis of the central nervous system, displaying some atypical features. This case highlights several key diagnostic and management issues of the disorder as well as its potential heterogeneity.
Keywords
Introduction
We report a biopsy-proven case of primary angiitis of the central nervous system (PACNS) with atypical features, particularly striking radiological asymmetry, and discuss important diagnostic and management issues of this rare disorder with specific reference to complexities highlighted by our case study.
Case report
A 62-year-old right-handed man presented with sudden dysphasia, presumed due to stroke. It later became apparent that he also had a poorly localised headache for 2 weeks prior without any other neurological symptoms.
He had type 2 diabetes on metformin and was a non-smoker who worked as a draftsman. On assessment, he was generally hyperreflexic with upgoing plantars but had normal power and sensation. He had a mixed dysphasia. Initial non-contrast computed tomography (CT) of the brain was normal; however, magnetic resonance imaging (MRI) revealed multifocal regions of diffusion restriction, almost exclusively involving the left parietal and temporal lobes. Unusually, for ischaemic stroke, there were also prominent areas of cortical fluid-attenuated inversion recovery (FLAIR) hyperintensity and thickening of the cortex (Figure 1). Magnetic resonance angiography (MRA) of the circle of Willis was normal. Repeat imaging 1 week later showed further progression with multiple new ischaemic lesions, extensive leptomeningeal enhancement restricted to the left hemisphere, particularly the left temporal lobe, as well as subcortical micro-haemorrhages and minor convexity subarachnoid blood which were not present on the first MRI (Figure 2). The abnormalities continued to be markedly asymmetric, almost exclusively involving the left hemisphere. Bloodwork, including full blood count, erythrocyte sedimentation rate, C-reactive protein and angiotensin converting enzyme was normal. Antinuclear antibodies were positive at a low titre of 1:80 with a nucleolar pattern, but an extensive battery of extractable nuclear antigens was negative. Serum results for antineutrophil cytoplasmic antibodies, rheumatoid factor, anti-cyclic citrullinated peptide, double-stranded DNA, IgG and IgM anticardiolipin antibodies, lupus inhibitor, dilute Russell’s viper venom time and cryoglobulins were all negative or normal. Results for tumour markers CA 125, CA 153, CA 199 and carcinoembryonic antigen (CEA) were also normal. Serology for HIV, hepatitis B, hepatitis C and syphilis was negative. Initial cerebrospinal fluid (CSF) was remarkable only for albuminocytological dissociation (protein 0.94 g/L with two mononuclear cells); however, 2 weeks later, a pleocytosis had evolved (white cell count 32, all lymphocytes with some cellular atypia) with further protein elevation to 1.75 g/L and the presence of xanthochromia. CSF polymerase chain reaction for herpes simplex virus, enterovirus, varicella zoster and Epstein–Barr virus was negative. Cryptococcus neoformans antigen titre was negative. Oligoclonal bands were not present.

(a and b) Axial DWI sequence MRI demonstrating scattered punctate ischaemic lesions in the left temporal lobe, arrows. (c and d) Axial FLAIR MRI showing multiple areas of cortical hyperintensity in the left hemisphere, block arrows. (e) Intracranial MRA demonstrating normal intracranial vessels.
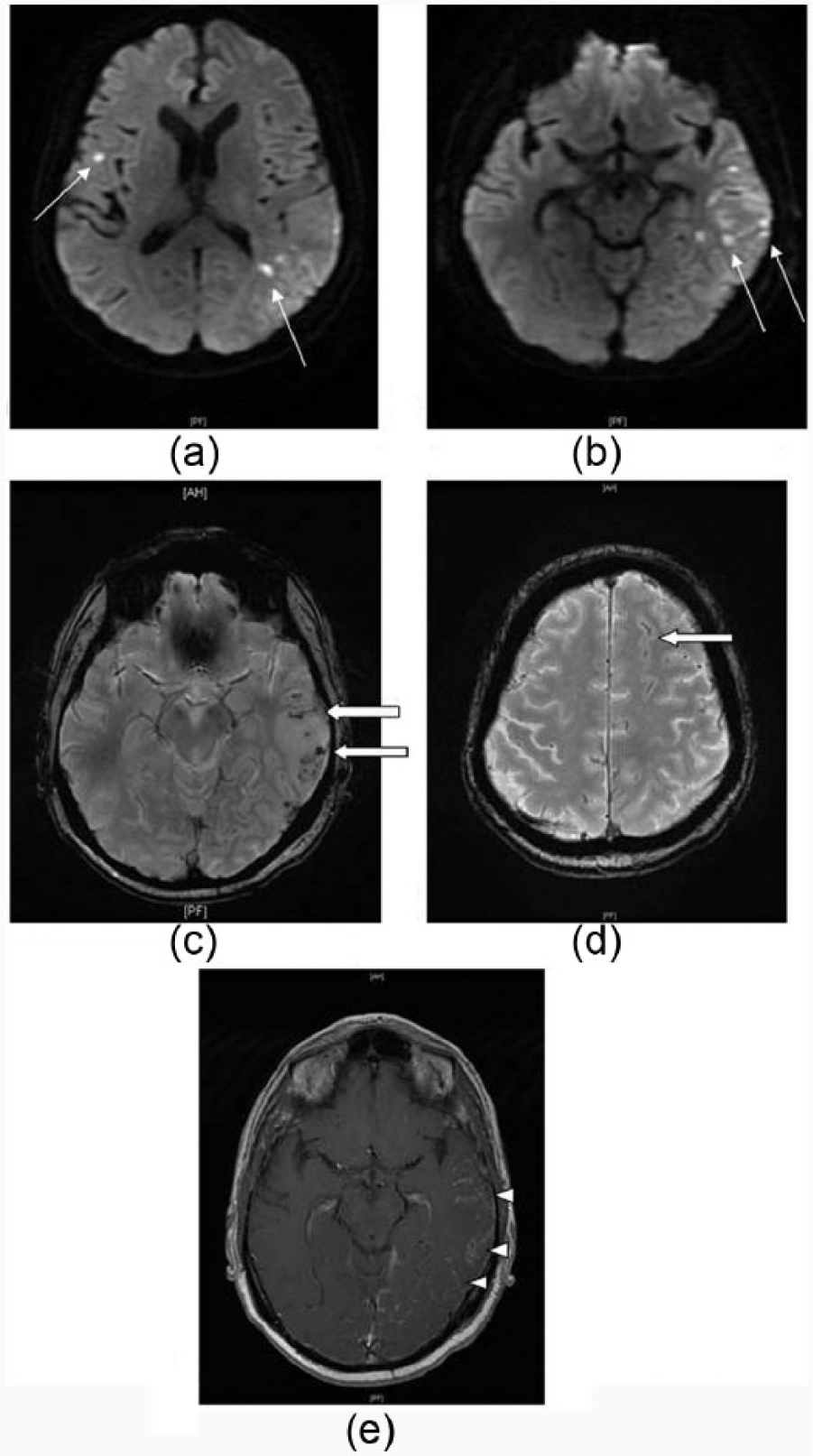
(a and b) Axial DWI MRI performed 9 days after Figure 1, revealing multiple new infarcts with left hemisphere predominance, arrows. (c and d) Axial gradient echo sequence showing multiple foci of increased susceptibility suggesting micro-haemorrhage, in addition to a linear area of subarachnoid haemorrhage in the left frontal region, block arrows. (e) Axial T1-weighted MRI with gadolinium showing leptomeningeal enhancement predominantly in the left temporal region, arrowheads.
Given the strong clinical suspicion of vasculitis, a meningeal and brain biopsy of radiologically abnormal tissue from the left inferior temporal lobe was undertaken. The findings were consistent with PACNS – showing patchy infarcts of variable age and vasculitic changes in small muscular cerebral and meningeal blood vessels consisting of transmural infiltrates predominantly of lymphocytes with fibrinoid change or infarction of the vessel walls (Figure 3). Congo red and crystal violet stains for amyloid were negative.

H&E stains from the left temporal lobe brain biopsy showing typical changes of PACNS. (a and b) Fibrinoid change in a superficial cortical small muscular blood vessel, arrow, as well as perivascular inflammatory infiltrate, predominantly lymphocytic with occasional histiocytes and multinucleated giant cells, block arrow, 40× and 200× magnification. (c and d) Area of cortical infarction, 40× and 100× magnification.
The patient was given 3 days of pulsed intravenous (IV) methylprednisolone (1 g/day) followed by oral prednisone (70 mg/day), complicated by significant destabilisation of glycaemic control and marked agitation and insomnia prompting relatively rapid steroid weaning by 10 mg weekly. Despite steroids, he continued to progress clinically and radiologically over 10 days, necessitating an induction course of oral cyclophosphamide (dose escalated over 1 week to 150 mg/day); favoured over the IV route due to difficult fluid balance management related to concomitant syndrome of inappropriate antidiuretic hormone secretion. He continued to progress radiologically with evolution of new infarcts over a further 3 months before eventual stabilisation, with slow clinical improvement and no new lesions on follow-up imaging at 12 and 18 months. Notably, although there were still many foci of T2/FLAIR hyperintensities as well as patchy susceptibility artefact in the left hemisphere, there were only ever a few, non-specific, small white matter changes in the right hemisphere.
Discussion
PACNS is a rare disorder, affecting an estimated 2.4 per million person-years with an average age at onset of 50 years and equal gender distribution. 1 It is a vasculitis of small- and medium-sized arteries, isolated to the brain, leptomeninges and sometimes spinal cord.
Diagnosing PACNS may prove challenging not only due to its rarity but also because of the variability and often non-specific presentations which necessitate a high index of clinical suspicion and rigorous investigation to confirm the diagnosis and exclude mimics. The presentation varies with the involved cerebral territory and may include headache, confusion, seizures and focal deficits. Disease onset is often insidious with slowly progressive symptoms, 1 often resulting in a prolonged delay between symptom onset and diagnosis. Less commonly, the presentation may be acute, with stroke or a transient ischaemic attack occurring in 30%–50% of patients. 2 Aphasia specifically occurred at presentation in 28% of patients in a large cohort. 1 Our patient’s abrupt symptom onset with an acute stroke syndrome, albei with a subtle brief preceding headache history, prompted aggressive investigation facilitating early diagnosis and treatment.
Imaging may be non-specific and the findings confused with other diagnoses. Helpfully though, as has been noted previously in the literature,1,3 the diagnosis is essentially excluded by a completely normal MRI (particularly if the CSF is also normal). Possible abnormalities seen with the disorder include multi-territorial infarcts, haemorrhages, FLAIR hyperintensity and meningeal or cortical contrast enhancement. While our patient’s imaging demonstrated most of these abnormalities, an unusual feature of our case was the striking and persistent asymmetry of disease, which was virtually confined to the left hemisphere, adding additional diagnostic complexity. In patients with PACNS and multiple infarcts, 83% have bilateral changes. 4 Less commonly, the disease may present with a tumour-like mass lesion. 5
Laboratory investigations are expected to be normal apart from the CSF which is abnormal in the majority of patients but again non-specifically, most commonly showing a mild pleocytosis and/or elevated protein. The utility of these analyses lies largely in excluding infective or neoplastic mimics, or secondary cerebral vasculitis due to a systemic autoimmune disorder.
Angiography (through either non-invasive techniques or formal digital subtraction angiography) classically reveals multifocal ‘beading’ with alternating stenoses and dilatations of medium-sized vessels but may be normal, especially in cases only involving small vessels. 6 There is significant variability in the reported sensitivity and specificity of angiography, with average sensitivity estimates of 60% in biopsy-proven series3,4,7 and specificity as low as 30%, 8 which highlights the difficulty of relying on imaging alone to prove or exclude the diagnosis. For these reasons, biopsy and histopathological analysis remain the diagnostic gold standard. Transmural vasculitis of small- to medium-sized leptomeningeal or parenchymal vessels is diagnostic. The vasculitic changes may be granulomatous, lymphocytic or necrotising. 9 Due to the segmental nature of the disease, negative biopsies may occur, further complicating the investigative process. This is less likely if radiologically abnormal tissue is biopsied, and both meningeal and cortical tissues are sampled 7 as in this case.
PACNS is an important differential to consider when diagnosing the more common reversible cerebral vasoconstriction syndrome (RCVS). 5 The presenting symptoms may be similar, and both can result in cerebral infarcts or haemorrhage, but a severe thunderclap headache at onset (which is often recurrent) should suggest RCVS. Angiography in this disorder is characterised by multifocal areas of reversible vasoconstriction which may appear worse than the clinical presentation suggests and which, crucial to the diagnosis, resolve on repeat imaging. The importance of differentiating the two entities lies primarily in the markedly different treatment paradigms. 5
While previously believed to have a particularly poor prognosis, the outlook for PACNS is now brighter, with many patients reportedly responding to high-dose steroids, plus or minus cytotoxic agents. 4 To date, however, there is no randomised trial evidence to guide therapy, including whether IV or oral regimens are more effective, or when to add a cytotoxic agent, or which agent to use. Individual case reports include ad hoc use of various agents, most commonly cyclophosphamide based on its efficacy in other vasculitic disorders. The prognosis has been shown to be improved by early diagnosis and prompt treatment, 1 which again emphasises the importance of clinical diligence in establishing the diagnosis. Potential treatment complications, several demonstrated by our case, reinforce that diagnostic accuracy is critical prior to treatment. A further management challenge in this case was the initial persistence of disease progression despite pulsed steroids and cyclophosphamide. Postulated mechanisms for progression despite aggressive treatment in a paediatric cohort include residual thrombogenecity from recent inflammation or early treatment tapering; 10 however, little is known of the exact pathophysiology. The necessity for early steroid weaning in our case may well have been contributory and serves to highlight one of the therapeutic challenges of this disorder in which treatment toxicity must be carefully weighed against disease morbidity. Certainly, there is little evidence regarding third-line therapeutic options, although case reports suggest potential efficacy for tumour necrosis factor-alpha blocking drugs in resistant disease. 1
It has recently become clearer that PACNS probably represents a spectrum of disorders, and ideally, further clarification of clinicopathological variations may help to hasten diagnosis and guide tailored treatments for subgroups. For example, there is a group of patients with cerebral amyloid angiopathy (CAA), with vascular deposits of beta amyloid, who have coexistent granulomatous vasculitis, recently termed amyloid beta-related angiitis (ABRA). 11 These patients tend to be older than typical PACNS but younger than other CAA patients, often present more acutely with cognitive dysfunction or focal deficits, have T2 or FLAIR hyperintensities (which may be asymmetrical or mimic a mass lesion) plus findings suggesting CAA (such as microbleeds) on imaging and usually respond favourably to immunosuppression or even steroids alone.1,10 Our patient displayed many of these features, although his histopathology ultimately excluded ABRA. Another group of PACNS present with rapidly progressive disease, 1 are typically slightly older than the general cohort, usually have multiple bilateral infarcts, prominent angiographic abnormalities, poorer response to therapy and higher mortality, perhaps warranting more aggressive management.
In summary, we believe that our case of PACNS highlights some key diagnostic and management challenges presented by this rare disorder: in particular, the complexity of diagnosing a potentially heterogeneous disease with variability of clinical presentation and radiological findings, with marked asymmetry a notable atypical feature in our case; and the difficulties in managing a severe disorder requiring potentially toxic therapies in the context of a limited evidence base to guide therapeutic decisions.
Footnotes
Declaration of conflicting interests
The authors declare no conflict of interest in preparing this article.
Ethics approval
Not required by authors’ institution.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Patient consent
Written informed consent was obtained from the patient for publication of this article.