
Abstract
The clinical picture of immunomediator disorders of the central nervous system resulting from autoimmune or paraneoplastic processes is often represented by the limbic symptom complex or limbic encephalitis. The article gives a brief description of these conditions, allocated to a separate nosological group in 2007. The symptoms of limbic encephalitis include mental disorders and epileptic seizures of both convulsive and non-convulsive spectrum, up to epileptic status. Four clinical cases representative of different variants of limbic encephalitis are presented in this study, along with the discussion of epidemiology, differential diagnostics, and generally accepted patient management strategies. The diagnosis of limbic encephalitis was made on clinical grounds alone in three cases and on the presence of antibodies to N-Methyl-
Keywords
Introduction
Background
Encephalitis resulting from an autoimmune destruction of synaptic proteins, which play a crucial role in neuronal transmission and plasticity, is a developing group of immunomediator disorders of the central nervous system (CNS). Target antigens include N-methyl-
Epidemiology
The variant of autoimmune encephalitis with antibodies to NMDAr (anti-NMDAr encephalitis) is the most common and studied to date. In 2005, four women with ovarian teratoma were described having a syndrome of cognitive deficits, mental disorders, decreased level of consciousness, and hypoventilation. 7 Soon, specific autoantibodies to NMDAr were detected in these and another eight patients with similar neurologic symptoms. Subsequently, seven of the latter eight patients were diagnosed with ovarian teratomas. 1 Over the next 3 years, another 419 patients were identified, some of whom were children and adolescents with a similar symptom complex with or without an associated tumor. The discovery of this disease, called anti-NMDAr encephalitis, has changed the diagnostic approach to such diverse clinical conditions as catatonia, subacute memory impairment, convulsions, motor disorders, and LE. 8 Since 2007, the disease is regarded as a separate nosology.
The exact incidence of anti-NMDAr encephalitis is unknown. Based on the rapid accumulation of patient data and the growing number of reports, this pathology appears to be more frequent than any other known paraneoplastic encephalitis. A multicenter population-based prospective study of the probable causes of encephalitis in the United Kingdom revealed anti-NMDAr encephalitis in 4% of patients. 9 The disease was the second most common after acute disseminated encephalomyelitis among the immune-mediated encephalites. It was more common than all other antibody-associated encephalitis, including encephalitis with involvement of the potential-dependent calcium channels. According to J. Dalmau et al., 10 from 2011, who described more than 400 patients with anti-NMDAr encephalitis over a period of 3 years, the disease can be considered relatively frequent. Approximately 80% of people with anti-NMDAr encephalitis were women. More than half of the patients had an associated tumor, usually an ovarian teratoma, which can be mistaken for a benign cyst. The diagnosis of a related tumor depended on age, sex, and ethnicity. In general, teratomas were detected in Black women over 18 years of age. Only 5% of male patients over the age of 18 years had an associated tumor.10–12 In another study, 25 patients with ovarian teratomas had production of antibodies to NMDAr. 13 Tumors other than teratomas are uncommon: of more than 400 patients described by J. Dalmau et al., 10 only 7 (2%) had a different tumor. One case with neuroblastoma and one with Hodgkin’s lymphoma have also been reported.14,15 Secretion of NR1 antibodies by other tumors was confirmed only in one case of breast cancer. 10 Whether tumors other than teratoma are truly associated with LE or only accidentally coincide with it is not clear.
Clinical features
Clinically, the involvement of the CNS synapses can be manifested by a diverse symptomatic complex, including catatonia, psychosis, motor disorders, short-term memory disruption, and refractory convulsions. Therefore, these patients may initially present to various medical specialists. A characteristic feature of the manifestation of the disease is a certain “phasic” structure: prodrome (non-specific Upper respiratory tract infection-like symptoms; about 5 days), psychotic phases (emotional, cognitive impairments, schizophreniform symptoms; 2 weeks), active (catatonic state, mutism, akinesia), hyperkinetic (orolingual and athetoid dyskinesias, symptoms of autonomic instability), and the reverse development of symptoms (2–6 months).4,16
With the existing common pathogenetic basis of the considered autoimmune synaptic disorders and their clinical similarity, where the limbic symptomatic complex is the key manifestation, it is necessary to distinguish a number of features that are more or less characteristic of certain nosological forms. First, the anti-AMPAr encephalitis is characterized by acute limbic dysfunction, which manifests as a rule with significant psychological symptoms 2 and is often accompanied by epileptic attacks, with approximately 70% of this group diagnosed with a lung tumor, mediastinum, or thymus. 17 Second, the encephalitis associated with antibodies to the GABAB receptor is usually manifested by convulsive attacks in addition to the limbic symptom complex. 18 There is a background of small-cell or neuroendocrine lung malignancy in approximately 50% of cases. 19 Third, in patients with anti-LGI1 LE, approximately 40% of cases of cognitive deficits may be preceded by convulsions resembling myoclonus. 20 This encephalitis was previously associated with antibodies to potential-dependent potassium channels. However, it has recently been shown that LGI1 is a secreted neuron peptide interacting with presynaptic and postsynaptic receptors, and its mutations were associated with autosomal dominant lateral temporo-frontal epilepsy syndrome. 21 Finally, LE associated with anti-CASPR2 is of particular clinical interest, as there is a combination of symptoms of encephalitis and increased excitability of peripheral nerves (Morvan’s syndrome), which can lead to the initial diagnosis of an atypical form of motor neuron disease. Immune-related disorders such as myasthenia gravis with antibodies to acetylcholine or antibodies to muscle kinase (MuSK) may be present in some patients, which confirms the association of these conditions with different CASPR2 proteins, which in fact are the target antigens in these patients. Syndromes associated with CASPR2 antibodies can develop both with and without an associated tumor. Since anti-CASPR2 encephalitis responds to immunotherapy, it should be distinguished from other forms of LE.4,22
Methods of diagnostics
Magnetic resonance imaging (MRI) in anti-NMDA encephalitis demonstrates a hyperintensive signal in Fluid-attenuated inversion recovery (FLAIR) or T2 regimens in the cerebral cortex or cerebellum cortex, or in mediastinal regions in about 50% of cases. 10 Neuroimaging changes can also be detected in the corpus callosum and brain stem; 1 a weak or transient contrast of the soft medulla over the cortex of the cerebral hemispheres, cerebellum, and basal ganglia was also described. 11
Electroencephalographic (EEG) studies allow detection of both the specific EEG patterns of epileptic paroxysms, including the regional slow wave (SW) and disorganized (D) bioelectrical activity (BEA) of the brain, and the paroxysms themselves10,11 up to non-convulsive status epilepticus (NCSE). 22 One of the EEG phenomena described in LE is the three-phase wave (TPW). TPWs are often detected in instances of toxic, metabolic, or structural damage to the brain substance.23–26 Interestingly, they are also found in about 13.6% of LE cases, 27 particularly in NCSE. 28 There is an opinion that TPWs are formed in conditions of metabolic disturbances and represent a pattern of NCSE. 29 Beta-delta pattern (BDP, extreme “beta-delta brushes”) is another element that appears on EEG of 30% of patients and is considered pathognomonic of the anti-NMDAr. BDP is symmetrical, synchronous, and spreading to all regions of the brain. It appears as long episodes of SWs in the main δ-range, on which fast β-vibrations are superimposed.30,31
Cerebrospinal fluid (CSF) abnormalities are present in 80% of patients. 11 As a rule, there are mild or moderate lymphocytic pleocytosis, a slight increase in protein and oligoclonal antibodies.8,11
Laboratory diagnosis of anti-NMDAr encephalitis is confirmed by detection of antibodies to the subunits of the NR1/NR2 NMDA receptor in the majority of cases in the serum or CSF.11,32 In the course of treatment or in the late stages of the disease, in the absence of clinical improvement, the CSF antibodies remain elevated, indicating a greater correlation of the titer of CSF antibodies with the outcome of the disease.10,33
The most recent guidelines of diagnosis of autoimmune encephalitis at early stages are based on clinical neurological evaluation and standard diagnostic tests (e.g. MRI, EEG, CSF analysis), whereas detection of specific antibodies enables verification of diagnosis and guides treatment. 34
All cases presented below underwent an extensive workup, which included negative laboratory screening tests for endocrinologic, rheumatologic, and sexually transmitted diseases. Moreover, cases 1, 2, and 4 underwent an autopsy which likewise failed to identify any evidence of autoimmune or oncologic pathology.
Principles of therapy
Treatment of anti-NMDAr encephalitis should be based on immunotherapy and the detection and removal of teratoma. In most cases, the first line includes glucocorticoids (GCs), intravenous immunoglobulins (IV Igs), or plasmapheresis.11,12,32 Parallel assignment of IV Ig (0.4 g/kg/day) and methylprednisolone (1 g/day) for 5 days is preferred. In patients without a tumor or with a delayed diagnosis, additional second-line drugs (rituximab, cyclophosphamide, or their combination) are usually necessary.35,36 With surgical treatment, the neurologic response to tumor removal can sometimes be seen within hours, 37 similar to the effect of anesthetics with the NMDAr blockade. Spontaneous neurologic improvement has been reported, with more prolonged hospitalization and slow recovery.8,10,16
Outcomes
Anti-NMDAr-, anti-AMPAr-, and anti-LGI1 (limbic) encephalitis are regarded as potentially curable, with possible relapses. For example, in anti-NMDAr encephalitis, relapses occur in 20% to 25% of patients, depending on the time of initiation of therapy. Patients who have started anti-tumor therapy with or without immunotherapy within 4 months after the appearance of neurological manifestations of the disease had fewer neurological relapses and better recovery than patients without tumors or patients with tumors who started treatment later or were not treated at all. The probability of recurrence of encephalitis does not depend on the activity of the tumor and can remain elevated after months or years, even after successful treatment and the absence of tumor recurrence, increasing with errors in therapy or with its termination.7,11,38 With NMDA encephalitis, about 75% of patients recover or have mild complications, with severe disability and mortality occurring in the rest of the cases. 11
Case 1
A case of limbic syndrome characteristic of anti-NMDAr encephalitis associated with ovarian teratoma is retrospectively observed in a patient treated in our clinic in 2003. Patient K., a 20-year-old female, had 3 days of experience of an influenza-like state, including fever up to 37.8°C, headaches, sore throat, general malaise, and sleep disturbance (prodromal stage). One week later, she had sharply developed a schizophreniform state with agitation, motor stimulation, and hallucinations (stage of mental disorders). 16 Following hospitalization in the psychiatric ward and the initiation of sedative therapy with diazepam, she suffered from fluctuating consciousness during the day from stunning to stupor, an impaired verbal contact, apart from pronouncing individual sounds, and loss of control over sphincters. The status epilepticus presentation included passive supine position, mostly with the eyes open, stiff neck muscles, floating eyeballs, divergent strabismus, dissociation of responses to external stimuli, a non-localized delayed reaction to pain, and resistance to opening eyes, resembling the action of dissociative anesthetics–NMDA antagonists, such as phencyclidine or ketamine. There were also opercular hyperkinesis, choreoathetosis of the extremities more to the right, torticollis to the right, and impaired swallowing (stage of motor and vegetative disorders). Breathing was intact. In the CSF, there was a slight increase in the protein content (0.78 g/L) and predominantly lymphocytic pleocytosis (up to 187 × 106/L cells, 179 × 10/L were lymphocytes), no oligoclonal antibodies, and no antibodies to NMDAr were performed at that time. No brain lesions were recorded on the MRI of the brain, and EEG demonstrated the predominance of the SW and D-BEA, characteristic of the anti-NMDAr type of LE, without epileptiform discharges. The TPWs were observed in the leads F4-A2, C4-A2, and BDP in C3-A1 (Figure 1).
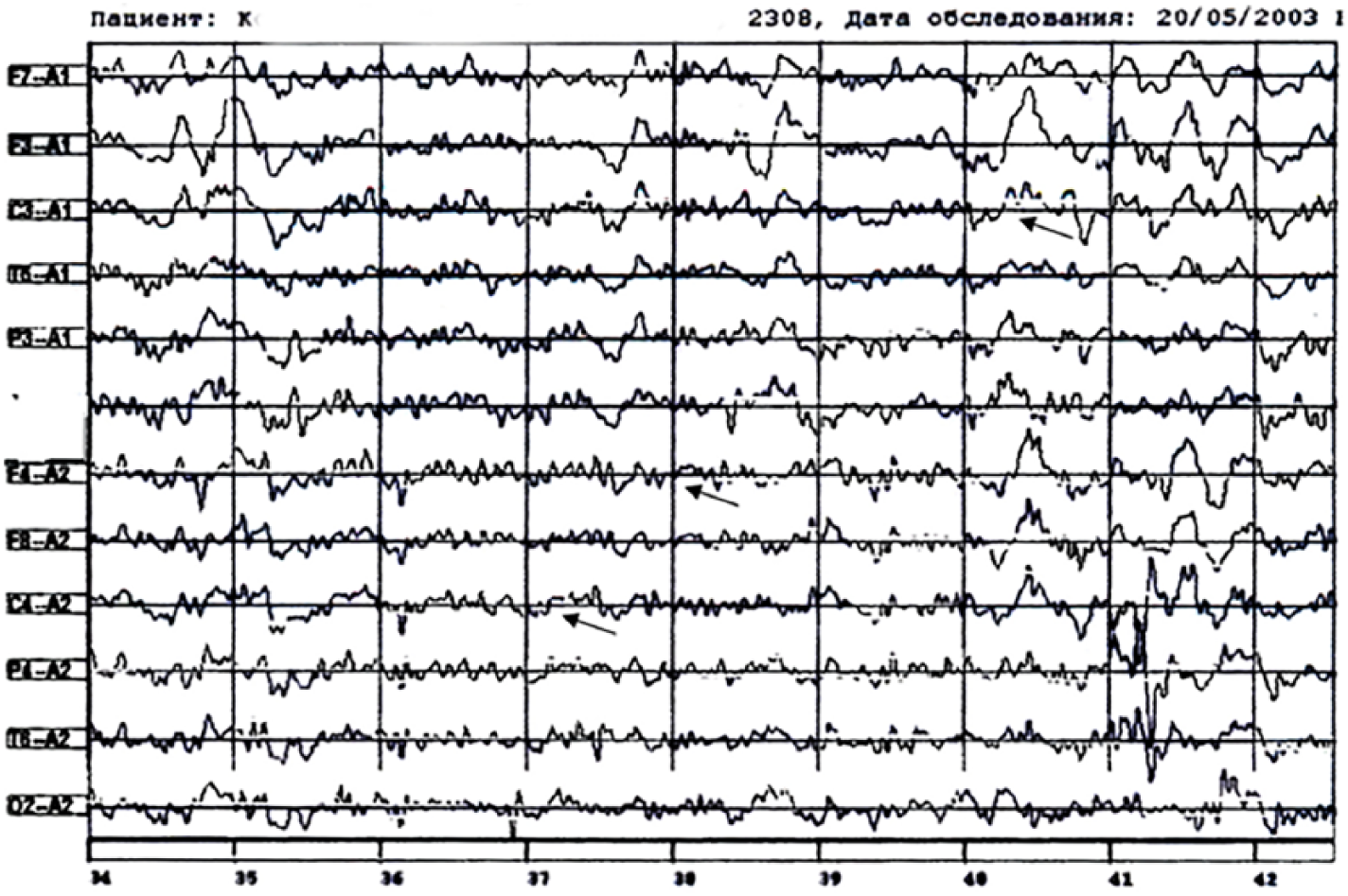
The EEG of patient K. (Case 1). Regional deceleration in central frontal parts, mostly on the left, triphasic waves on leads F4-А2, С4-А2, beta-delta brushes on lead С3-А1; state of consciousness: stunned.
A teratoma of the right ovary was found, regarded at that time as an unrelated finding. A differential diagnostic search was carried out, aimed at ruling in tuberculosis, rabies, and the use of psychotropic and narcotic drugs; none of the above was confirmed. As a result, the disease was defined as viral lethargic encephalitis. Within 3 weeks, the patient received posidrome, antibiotics, and immunotherapy (human Ig). GCs were not used, as there were no data on the presence of actual autoimmune pathology. There were some positive dynamics on days 12 to 14—an increase in activity, recognition of relatives, improvement of speech (pronounced separate syllables and simple words), and the patient began to perform simple commands. The patient had suffered from lethal thromboembolism to the pulmonary artery after 3.5 weeks of treatment.
Case 2
Patient S., a 55-year-old female, presented, 31 January 2014, with inhibition, disorientation, decreased motor activity (preferred bed rest), and an impaired verbal contact (became monosyllabic when talking and answering questions). Other symptoms included dizziness, headache, and thirst. In view of the existing concomitant diseases—untreated arterial hypertension and obesity—an acute stroke was suspected, which was not confirmed on the same day by brain computed tomography. For 1 month, she was on outpatient treatment with a working diagnosis of “decompensation of chronic cerebral circulatory failure,” with a gradual restoration of functions to the initial satisfactory state. After 1 week of general malaise and hyperthermia, she was found by relatives unconscious, with foam at the mouth, probably after a seizure attack. After the ruling out stroke, she was hospitalized in an internal medicine ward, where after 2 days the consciousness recovered to the level of stunning, and clear by the end of the first week of therapy. On the 14th day, she was discharged home in a satisfactory condition. During the following month, after a period of normal function (she was serving herself, went to the store), a relapse of deterioration began on 23 March 2014, similar to the episode of 31 January 2014, and then—on 4 April 2014—the patient sustained a bilateral tonic–clonic seizure (BTCS), which resulted in a second hospitalization. After discharge on 10 April 2014, the patient answered monosyllables for questions, was unable to ambulate, and could not perform activities of daily living independently. In the following days, relatives noted the appearance of twitching of the muscles of the face and limbs, which was the reason for hospitalization on 17 April 2014 in the neurological department. At the time of admission, the condition was severe, the level of consciousness was moderate stunning, and the patient did not contact (did not do the commands, did not speak) and did not react to the examination.
Active movements in all limbs were preserved. Myoclonus was noted in the face, both arms, and right leg. On the fourth day of anticonvulsant (valproic acid (VPA): 1000 mg/day), neurometabolic, and hypotensive therapy, the patient began to talk partially and to sit, and the Mini-Mental State Examination (MMSE) showed 20 points. In the next 2 days, S. began to walk independently, to answer questions on a limited scale, to carry out simple instructions, and to control the sphincters more often. Against the backdrop of further positive dynamics of the patient’s condition, inoculation and disorientation recurred on 6 May 2014, and the next day laryngeal edema and a series of BTCS developed, requiring transfer to the intensive care unit. The seizures were stopped on the same day, and the edema of the larynx (attributed to infectious-allergic pathology) was relieved within 7 days with 12 mg/day dexamethasone. Since 24 May 2014, the appearance of hallucinatory-delusional disorders has been documented: the patient claimed to be at home, washed floors in other wards, complained on neighbors all plotting against her. Since 31 May 14, pulse therapy with methylprednisolone (1000 mg for 6 days) was started, after which there was a positive dynamics in the form of stopping hallucinations, restoring productive verbal and motor activity. In the process of examination in a hospital, the patient excludes topical oncological and dysmetabolic processes; there was also no pathology of the brain (MRI). On the EEG we observed polyrhythmic SW activity without epileptiform phenomena, and also BDP in leads F3-A1, C3-A1, P3-A1, O1-A1 (Figure 2). In CSF, protein: 1.02 g/L, normal cell count (29 May 2014); antibodies to NMDAr were not detected. S. was discharged from the department on 11 June 2014 in a satisfactory condition (had put on a makeup herself); EEG: normal variant (Figure 3) and MMSE test: 30 points. Further therapy: Valproic acid at a dose of 1000 mg/day; per os for a long time; GC was not prescribed.
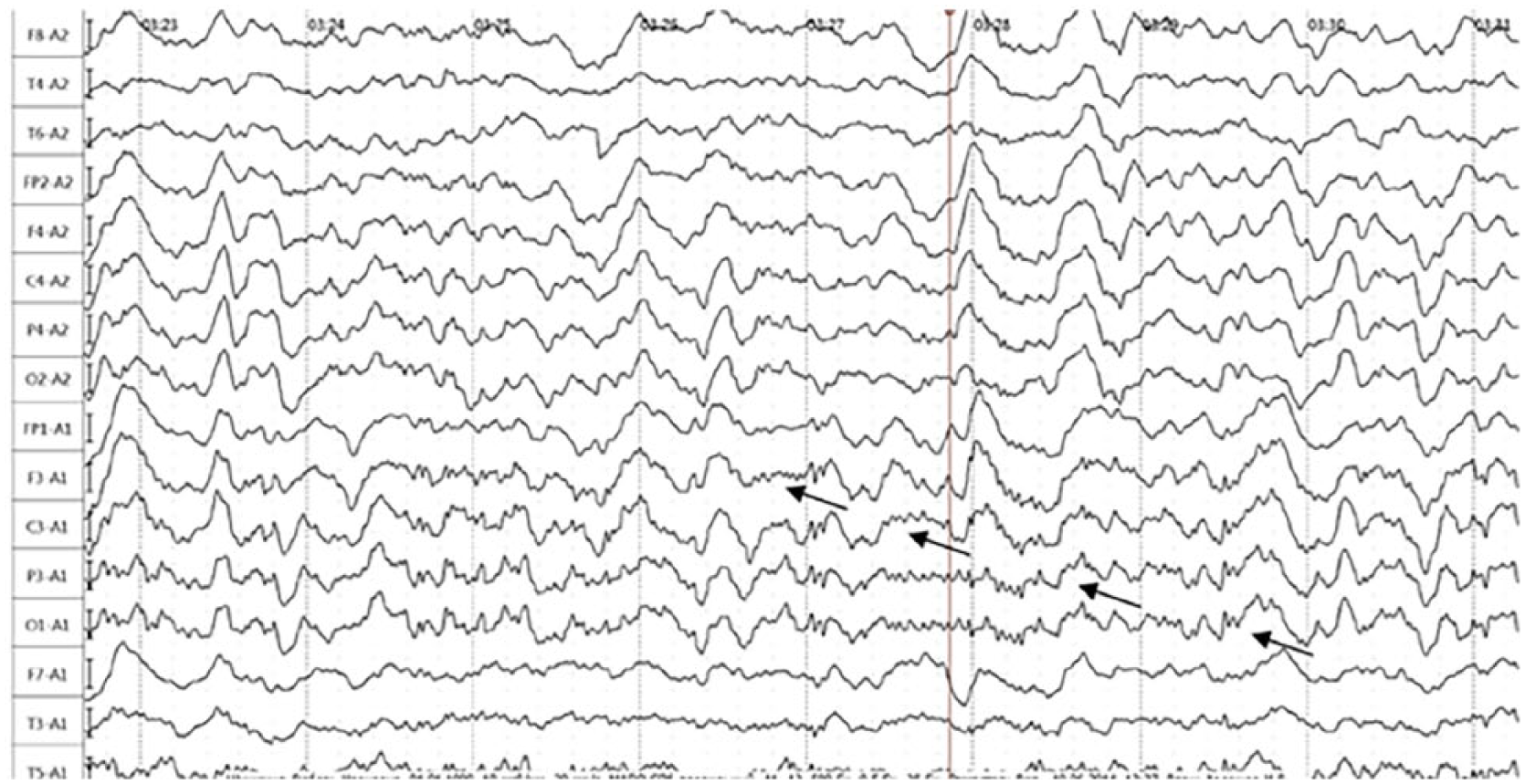
The EEG of patient S. (Case 2), made 1 day prior to commencement of generalized tonic–clonic seizures; beta-delta brushes on leads F3-А1, С3-А1, Р3-А1, and О1-А1 (arrows); state of consciousness: stunned.

The EEG of patient S. (Case 2). Normalization of the EEG; inter-paroxysmal period, state of consciousness: lucid.
Case 3
Patient Z., 71-year-old male, presented with a 2-week complaint of general malaise, increased fatigue, mild headache, and episodes of short-term dizziness not complicated by fall. According to relatives, memory for current events decreased, and short-term (up to a minute) episodes of loss of consciousness appeared (up to several times a day), during which the patient froze and did not come into contact with others. In addition, the difficulties in communication intensified with the appearance of periods of reasoning. The general malaise and dizziness had progressed, and a hypertensive crisis on 6 February 2018 led to admission to the internal medicine department with a diagnosis of cerebrovascular disease. The condition did not improve after 2 weeks of symptomatic therapy, and the patient was transferred to outpatient treatment. When the MRI of the head was performed in the FLAIR mode, a lesion in the medial parts of the left temporal lobe was detected (Figure 4). The patient was consequently hospitalized in the neurological department on 29 March 2018 with the alleged diagnosis “limbic encephalitis.” On admission, the condition was relatively satisfactory, the consciousness was clear, the understanding of reversed speech was difficult, and there were exaggerated tendon and periosteal reflexes with the expansion of reflexogenic zones, Rossolimo’s sign positive bilaterally, and easily expressed instability in Romberg’s position. CSF contained a significantly increased protein content (1.1 g/L), erythrocytes (250 × 106/L cells), and predominantly lymphocytic pleocytosis (up to 23 × 106/L cells); no antibodies to NMDAr were detected. Herpetic infection (IgM, IgG) was ruled out. The EEG (30 March 2013) demonstrated a periodic regional slowdown in the leads Fp1-F7 (Figure 5). At the same time, in the contralateral area (leads Fp2-F8), a regional BDP (beta-delta brush) was recorded (Figure 6). In the period of hospital stay—5 April 2018—there was a single BTCS, which entailed the appointment of VPA in a dose of 600 mg/day per os. In the repeated EEG from 6 April 2018, the marked changes regressed (Figure 7). After treatment with GC (methylprednisolone) IV and plasmapheresis, the patient’s well-being improved, the symptoms completely regressed, and the attacks did not recur. The patient was discharged home for outpatient follow-up with recommendation to continue treatment with VPA.

Head MRI of Patient Z. (Case 3). An irregular intensification of signal in left temporal lobe in regime FLAIR (white arrows).

The EEG of patient Z, 30 March 2018 (Case 3). Periodic regional decelerations in the left rear frontal (anterior-temporal) lobe (leads Fp1-F7 (arrows)). Consciousness level: altered, eternally observed as passive awakening.

EEG of patient Z., 30 March 2018 (Case 3). Regional beta-delta pattern in right rear frontal (anterior-temporal) lobe (leads Fp2-F8 ((arrow)).

EEG of patient Z., 6 April 2018 (Case 3). Regression of epileptiform patterns after treatment with valproic acid. State of consciousness: lucid.
Case 4
Patient K., a 17-year-old male, on 24 July 2016, noted the sudden numbness of the left arm, then of the right leg, after which a BTCS developed with a short postictal period in the form of generalized muscle weakness, speech fuzziness, and disorientation. A repeated BTCS occurred in several hours, manifested by the clone of the left hand and loss of consciousness. When hospitalized in a children’s neurological department with a diagnosis of “focal epilepsy,” disorientation in time and place, inadequacy of behavior, motor automatisms in the form of biting lips, waving hands, and attempts to remove clothing were noted. During hospitalization, a partial amnesia of events was observed, and the patient experienced repeated episodes of motor activity and inadequate behavior (he ran around the department, pushed medical personnel, and did not react to remarks). The episodes of an increase in body temperature to 37.6°C and an elevated blood pressure—up to 150/90 mm Hg—were recorded. The patient was transferred to a general psychiatric department on 1 August 2016 with a preliminary diagnosis of “focal epilepsy, psychosis.” The episodes of confused consciousness accompanied by psychomotor agitation and motor stereotypes persisted. In the periods between paroxysms, the patient experienced confusion, episodes of catatonia, and negativism. Progression of vegetative disorders progressed in the form of an increase in blood pressure to 150/100 mm Hg, heart rate to 126 beats/min, subfebrile body temperature, severe sweating, pale skin, and blood glucose increase up to 160 mg/dL. The paroxysms were amnesed by the patient. There were no delusional hallucinatory disorders. As part of the diagnostic search, EEG was performed three times and was invariably disorganized with predominance of alpha activity, and periodic regional slowdown together with regional BDP in the left frontal parietal region. Head MRI was performed twice, including with contrast enhancement, failing to visualize any pathology. CSF (3 August 2016) analysis showed a slight increase in protein (0.53 g/L) and very slight lymphocytic pleocytosis. Assessment of the internal organs (ultrasound, MRI) has not revealed pathology. After starting therapy with haloperidol, a negative symptomatology developed within 1 day, including appearance of muscular dystonia. The patient was transferred to an intensive care unit with a diagnosis of “febrile attack of schizophrenia.” Following a proposed diagnosis of autoimmune encephalitis, GC (methylprednisolone 1000 mg IV/cap. no. 6) was administered. Due to the ineffectiveness of treatment (symptomatic progression) and the detection of antibodies to NMDAr (1:160 titer) in the CSF and serum, the patient was transferred to the neurological department on 24 August 2016 in order to start plasmapheresis. Upon entering the department, the patient’s condition was regarded as severe (hyperthermia (39°C), consciousness—stunning, constant oromandibular dystonic hyperkinesis, muscle hypertonia). Despite plasmapheresis and symptomatic therapy, there was a progressive impairment of consciousness and breathing (with transfer to mechanical ventilation); on 29 August 2016, the death of the patient occurred.
Discussion
Despite some differences in the presented clinical cases, their common base is typical, as previously described, manifestation of the disease with acute limbic symptomatology, which included a phasic course characteristic of LE and paroxysmal impairment of consciousness belonging to epileptic spectrum.7,8
We consider the first clinical case to be a paraneoplastic process, clinically realized in the form of anti-NMDA-associated encephalitis associated with the teratoma of the ovaries.1,10 Unfortunately, the teratoma was not regarded as a condition associated with a clinically manifested disease; as in 2003, there was no theoretical justification for the possibility of such a connection, and a key analysis in the diagnosis of CSF for antibodies to NMDAr was not available at that time.
In the second clinical case, the phasic character of the process4,16 was less clear-cut and there was no tumor. However, other manifestations of the disease, including psychotic and paroxysmal, and both convulsive10,11 and non-convulsive symptoms, 28 were represented significantly. With a high degree of probability, the disease has an autoimmune nature, which is confirmed by a significant improvement and stabilization of the patient’s condition at the optimal functional level after the inclusion of GC in the therapy regimen.32,35,38 The second case is therefore distinguished by the correct identification and treatment of the limbic symptom complex from the start, and by the purposeful search for its cause and use of immunosuppressive therapy.
The third clinical case is of interest primarily because of the focal changes in the projection of the limbic system, visualized by the head MRI. These changes were observed in about half of the patients with LE. 10 This had enabled the diagnosis of the process and effective treatment of the patient in a time-limited clinical symptomatology.
The fourth clinical case demonstrated a combination of the phasic character of development of the clinical picture characteristic of NMDAr encephalitis with laboratory confirmation of the process.11,32 However, other typical paraclinical data did not confirm the final diagnosis. As a feature, the manifestation of the disease should be considered motor epileptic seizures in conjunction with severe mental disorders that had suddenly developed in an otherwise healthy subject.
It is worth mentioning the negative point that unites the above clinical cases: the diagnosis and the appointment of adequate therapy were delayed, which could undoubtedly affect the adverse outcomes of the disease.
Conclusion
Described cases emphasize the diversity of clinical presentation of LE. Physicians of various specialties who manage patients with autoimmune disorders of the nervous system should be aware of this diversity. LE should be suspected in any case of impaired consciousness of unclear etiology, and the patients should receive a complete workup.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.