
Abstract
Objectives:
Understanding the role of TRIB3 in cellular chemotherapy responsiveness and survival could facilitate its development as a prognostic marker that could be used to improve chemotherapeutic efficiency against specific tumors. Therefore, the role of TRIB3 to reflect the cytotoxic abilities of chemotherapeutic agents was clarified in the tested gastric cancer cell lines.
Methods:
We have comprehensively investigated the protein expression of TRIB3 in three gastric cancer cell lines AGS, TMK-1, and MKN-45 cells treated with the anticancer drugs, 5-fluorouracil, cisplatin, and docetaxel. The Cell Count kit-8 was used to evaluate cell viability. Immunoblotting was performed to assay protein levels after drug treatment. Flow cytometry was carried out to evaluate the levels of sub-G1 cell population.
Results:
Treatment of the tested gastric cancer cell lines dose-dependently decreased cell viability and protein levels of TRIB3 while increasing apoptosis. Overexpression of TRIB3 protects MKN-45 cells from endoplasmic reticulum stress-induced apoptosis but does not influence the induction of autophagy by anticancer drugs. In addition, overexpression of TRIB3 also rescued paroxetine-induced apoptosis and endoplasmic reticulum stress.
Conclusions:
Our previous and present results indicate that TRIB3 can protect gastric cancer cells against anticancer drug treatment and that downregulating TRIB3 may increase these cells’ sensitivity to anticancer drugs. We thus suggest that the capability of anticancer drugs to downregulate TRIB3 can indicate tumors’ potential susceptibility to these drugs.
Introduction
Gastric cancer has some of the highest prevalence and mortality rates among malignant tumors. Stomach cancer, including gastric cancer/gastroesophageal junction cancer (GC/GEJC), is the fifth most common cancer type and the fourth leading cause of cancer-related death globally, accounting for approximately 1 million new cases and 769,000 deaths in 2020. 1 In East Asia, gastric cancer is one of the most common malignancies. The standard of care for resectable GC/GEJC in Western countries consists of neoadjuvant-adjuvant 5-fluorouracil, leucovorin, oxaliplatin, and docetaxel chemotherapy combined with surgery. 2 Although treatment advances have improved survival, the prognosis of patients with gastric cancer remains suboptimal, with a 5-year overall survival (OS) rate of 33% in the USA and 25% worldwide. 3 New treatment options include the use of anti-angiogenesis and immune checkpoint inhibitors. Although the mortality rate of gastric cancer has been reduced, this disease still has a poor prognosis and a high worldwide mortality rate (second only to that of lung cancer). Predictive biomarkers have been proposed as a means to help refine post-surgery treatment selection and move the field toward precision medicine. However, new prognostic factors are needed to help refine therapeutic approaches and develop more effective therapies with reduced side effects.
5-Fluorouracil (5-FU), which is commonly used against human tumors, forms the backbone of most regimens used for gastrointestinal tract cancers. Mechanistically, 5-FU is a pyrimidine antimetabolite (fluorinated analog of uracil). There are three principal cytotoxic metabolites of 5-FU: 5-FdUMP (fluorodeoxyuridine monophosphate), which inhibits thymidylate synthetase and can damage DNA; FdUTP (fluorodeoxyuridine triphosphate), which along with FdUDP (fluorodeoxyuridine diphosphate) is generated from the intermediate metabolite FUDP, and can be misincorporated into and damage DNA; and FUTP (fluorouridine triphosphate), which can misincorporate into and damage RNA. Currently, 5-FU is used alone or in combination with other agents, such as oxaliplatin or irinotecan. It can also be combined with anti-angiogenic and/or anti-epidermal growth factor monoantibodies in treating human cancers, including gastric cancer.4,5 However, the responses to 5-FU-based chemotherapies can vary among individuals and 5-FU resistance represents a major issue constraining the efficacy of 5-FU in the clinic. Some biomarkers for predicting treatment responses in cancer patients have been investigated, but their clinical applications remain unclear. Thus, it is imperative to investigate novel biomarkers that can predict chemosensitivity and help tailor treatments to individual patients. Administration of standard 5-FU leads to severe side effects in 15%–30% of cases, and lethal toxicities are regularly reported with fluoropyrimidine drugs. Toxicities associated with 5-FU overdoses include hematological toxicities, gastrointestinal effects, cardiotoxicity, and neurotoxicity. 5-FU poisoning is influenced by several factors, including circadian rhythms, possibly gender, drug interactions, and genetic mechanisms. Thus, novel and safe treatment strategies that can overcome chemoresistance, diminish life-threatening toxicity, and enhance cancer responsiveness to 5-FU-based chemotherapies are desperately needed. Several relevant strategies have been explored, such as developing new 5-FU derivatives, using 5-FU-conjugated nanoparticles to maintain a higher prolonged 5-FU concentration in blood and tumor tissues, or combining treatment with other chemicals to sensitize tumor cells.
The taxane derivative, docetaxel, is an effective chemotherapeutic agent against multiple solid tumors, including breast, lung, ovarian, bladder, prostate, and head and neck cancers. Mechanistically, docetaxel promotes microtubule assembly by binding to the β subunits of tubulins and also inhibits depolymerization; this disrupts tubulin dynamics and inhibits mitotic spindle assembly at M phase. 6 Since critical cellular processes require the dynamic reorganization of microtubules, docetaxel treatment can inhibit major cellular events, including mitotic cell division, endosomal uptake, secretion, and transport. Docetaxel also inhibits vascular endothelial growth factor (VEGF) and exerts immunomodulatory and pro-inflammatory effects by inducing various mediators of the inflammatory response. It has been studied for use as a radiation-sensitizing agent 7 and can affect many signaling and molecular pathways involved in cancer cell survival and growth. Ultimately, docetaxel-treated cells undergo cell cycle arrest and apoptosis. 6 In addition to prostate cancers, where docetaxel is the most common first-line treatment and benefits OS, docetaxel-based chemotherapy can also be used in gastric cancer. For example, docetaxel given in combination with cisplatin increases OS in gastric cancer patients. 8
Cisplatin is a small and remarkably simple molecule composed of one platinum atom linked to two amides and two chlorides. It represents one of the most powerful chemotherapeutic drugs and is widely used to treat many types of cancer. Under low-chloride conditions, such as those found in the cytosol, cisplatin undergoes “aquation,” wherein one or two chlorides are replaced with water molecules. Aquation causes cisplatin to become highly reactive and able to readily bind various biomolecules inside the cell. 9 Reactive cisplatin largely targets DNA bases to form DNA adducts. The main action points for this are the nucleophilic N7-sites of purine bases, where a double reaction may covalently link purines. Cisplatin exerts its fundamental anti-neoplastic activity by binding to DNA and inducing DNA intrastrand and interstrand crosslinks in living cells. 10 Cisplatin forms primarily 1, 2-intrastrand crosslinks (90%) between adjacent purines in DNA, along with 1, 3-intrastrand crosslinks (5%–10%) and interstrand crosslinks (1%–2%). Nucleotide excision repair is largely responsible for repairing cisplatin-induced intrastrand crosslinks, whereas homologous recombination, translesion synthesis, and nucleotide excision repair are involved in repairing cisplatin-induced interstrand crosslinks. Cisplatin-induced DNA crosslinks and adducts strongly inhibit DNA replication and gene transcription, thereby activating multiple signal transduction pathways and inducing cell death. 10 However, although cisplatin shows broad-spectrum anticancer activity, its clinical utility has been limited by issues with acquired drug resistance, serious side effects, and damage to non-targeted tissues. Long-term off-target effects induced by chemotherapeutic drugs are a major cause of mortality among cancer survivors in later stages of life.
TRIB1, TRIB2, and TRIB3 are the human homologs of an evolutionarily conserved pseudokinase; they use non-catalytic mechanisms to fundamentally regulate the cell cycle, differentiation, metabolism, proliferation, and stress responses. TRIB3 is upregulated by hypoxia, endoplasmic reticulum (ER) stress, or nutrient deprivation and acts as a scaffolding subunit and modulator of several signaling pathways. 11 In recent decades, multiple studies have documented that TRIB3 contributes to apoptosis via the induction of ER stress, although some controversy remains. For example, TRIB3 is reportedly induced by various forms of ER stress. It contributes to CAAT/enhancer binding protein (C/EBP)homologous protein (CHOP)-dependent cell death under ER stress, and TRIB3 knockdown dramatically attenuates ER stress-dependent cell death. 12 In contrast, however, TRIB3 was reported to play a critical role in sensing growth inhibitory signaling and nutrient depletion and providing survival signals against these stresses.13,14 Consistent with the latter findings, we previously reported that doxorubicin transcriptionally downregulates TRIB3 to trigger apoptotic cell death, and knockdown of TRIB3 enhances doxorubicin-induced cytotoxicity. 15 Aberrant regulation of TRIB3 has been implicated in the etiology and progression of various diseases, including neurodegenerative diseases and cancers. There is some debate regarding the role of TRIB3 in tumor progression, as TRIB3 has been reported to be both increased and decreased in cancer patients compared with control groups. A tumor-promoting activity for TRIB3 is supported by reports that TRIB3 expression is elevated in patients with colorectal, breast, renal cell carcinoma, and non-small cell lung cancer, and its expression correlates with poor prognosis and OS in these patients. 11 However, tumor suppressor activity has also been reported some cellular and animal models of cancer, where TRIB3 promotes oncogene transformation and enhances the tumorigenicity of cancers. 16 Most of the recent studies focus on the correlation of TRIB3 expression with cancer malignancy, migration ability, OS rate, or immune therapy response.17–20 These studies all point to abnormal regulation of TRIB3 transcription, translation, and/or protein turnover as driving disease, but the underlying mechanisms, including the specific cellular functions and disease-associated signaling pathways, have not been evaluated in depth.
To improve targeted cancer therapy, chemosensitivity prediction, and/or the ability to tailor treatments for individual patients, additional potential biomarkers must be explored and implemented. It is generally accepted that 30%–80% of the severe toxicities recorded after 5-FU intake could be attributable to impaired dihydropyrimidine dehydrogenase (DPD) activity in the liver, and thus pre-treatment monitoring of DPD status could guide adaptive dosing strategies to reduce the incidence and severity of side effects. 21 Given our previous observation that TRIB3 is downregulated at the mRNA and protein levels when gastric cancer cells are treated with doxorubicin 15 and a report that the TRIB3-encoding gene is upregulated in the nano-Au particle-response signature, 22 we hypothesized that TRIB3 could play a critical role in the ability of cancer cells to survive under various stresses. Here, we comprehensively investigated the protein expression of TRIB3 in three gastric cancer cell lines AGS, TMK-1, and MKN-45 cells treated with the anticancer drugs, 5-FU, cisplatin, and docetaxel, and assessed whether TRIB3 could reflect the cytotoxic abilities of these chemotherapeutic agents in the tested cell lines. Our present data provide an alternative suggestion that TRIB3 might be another potential predictive biomarker for anticancer agent sensitivity and a potential therapeutic target in gastric cancer.
Materials and methods
Chemicals and antibodies
The protease inhibitor cocktail was a product of Roche Applied Science (Mannheim, Germany). 5-FU, cisplatin, and docetaxel were purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were purchased from Sigma-Aldrich or Amresco (Solon, OH, USA). Antibodies were obtained against the following: TRIB3 (Atlas Antibodies, Bromma, Sweden), p62 (Sigma-Aldrich), LC3, PARP, caspase 3, phospho-PERK, phosphor-eIF2α, Bax, Bim (all from Cell Signaling Technology, Beverly, MA, USA), and beta-actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The peroxidase-conjugated secondary antibodies against mouse and rabbit IgG were from Jackson Immuno Research Laboratories (West Grove, PA, USA). JetPEI (Polyplus, Illkirch, France) was used to transfect expression plasmids into cells according to the manufacturer’s recommendations.
Cell culture
The human gastric carcinoma cell lines AGS, TMK-1, and MKN-45 were cultured in RPMR-1640 supplemented with 10% fetal bovine serum and penicillin/streptomycin (all from Invitrogen, Carlsbad, CA, USA), at 37°C in a humidified 5% CO2 incubator. The AGS (accession number CVCL_0139) and TMK-1 (accession number CVCL_4384) were kindly gifted by Dr Chun-Ying Wu (Taipei Veterans General Hospital, Taipei, Taiwan). The AGS cell line was purchased from the Bioresource Collection and Research Center (BCRC; Hsinchu, Taiwan). The DNA short tandem repeat profile of the TMK-1 cell line was confirmed at BCRC. The MKN-45 cell line (accession number CVCL_0434) was purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan).
Cell viability assay
Cells (5 × 103/well) were seeded in a 96-well plate, incubated for 24 h, and treated with different concentrations of 5-FU, cisplatin, and docetaxel (three wells per concentration) for 48 h. Cell viability was detected using the Cell Count kit-8 (CCK-8; Dojindo Molecular Technologies, Rockville, MD, USA) reagent according to the manufacturer’s recommendations.
Immunoblotting analysis
Treated cells were collected and lysed in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 50 mM NaF, 1 mM Na3VO4, and 10% glycerol plus a protease inhibitor cocktail) at 4°C for 30 min. Equal amounts of proteins were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Immobilon polyvinyldifluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat dry milk for 1 h at room temperature and then probed with primary antibodies (1:1000) at 4°C overnight. The blots were washed and then incubated with peroxidase-conjugated secondary antibody (1:10000) for 1 h at room temperature. The signals were developed using an enhanced chemiluminescence system (Millipore). The images shown are representative of at least three independent experiments carried out under the same conditions.
Flow cytometry
Cells cultured in 6-cm dishes were trypsinized and collected by centrifugation. Each cell pellet was washed, resuspended in phosphate-buffered saline, and stained with 50 µg/ml propidium iodide (PI; Sigma-Aldrich) and 20 µg/ml RNaseA (Sigma-Aldrich). The distribution of sub-G1 cells was analyzed using a Beckman Coulter FC500 (Beckman Coulter Inc., Brea, CA, USA). The results are expressed as the percentage of total cells. For the detection of acidic vesicles in cells, each cell pellet was washed and stained with acridine orange (1 µg/ml; Sigma-Aldrich) for 15 min at 37°C. The images shown are representative of at least three independent experiments carried out under the same conditions.
Statistical analysis
All data were obtained from at least three independent experiments. Statistical calculations were performed using SigmaPlot 12.5 software (Grafiti LLC, Palo Alto, CA, USA). Values are presented as the mean ± SD from at least three independent experiments unless otherwise indicated. For statistical analysis, each experimental value was compared to its corresponding control. The statistical significance of differences between mean values was estimated using the t-test. The results presented in the graphs are representative of multiple independent experiments, and * indicates p < 0.05, which was considered statistically significant.
Results
Anticancer drugs attenuate TRIB3 protein expression and induce apoptosis
To comprehensively assess the effect of conventional anticancer drugs on gastric cancer cells, we first treated AGS, TMK-1, and MKN-45 cells with different concentrations of 5-FU, cisplatin, and docetaxel for 48 h. Cell viability was measured (Figure 1(a)–(c)), and IC50 values were calculated (Figure 1(d)). The cell viability of all the gastric cancer cells was dose-dependently decreased upon the treatments of 5-FU, cisplatin, and docetaxel (Figure 1(a)–(c)). We then treated AGS, TMK-1, and MKN-45 cells with 0.5 IC50, 1 IC50, and 2 IC50 of 5-FU, cisplatin, and docetaxel for 48 h and assessed apoptosis and TRIB3 protein expression. Consistent with our previous results obtained with doxorubicin, 15 the protein level of TRIB3 was dose-dependently decreased while apoptosis (assessed by detecting the cleaved forms of PARP and caspase 3) was dose-dependently increased by 5-FU, cisplatin, and docetaxel treatment of the tested gastric cancer cell lines (Figure 2(a)–(c)). Flow cytometric analysis of the sub-G1 cell population (Figure 3(a)), which is another indicator of apoptosis, confirmed that 1 IC50 of 5-FU, cisplatin, or docetaxel significantly enhanced the fraction of sub-G1 cells (and therefore apoptosis) in all three cell lines (Figure 3(b)). Our previous and present data therefore indicate that conventional anticancer drugs downregulate TRIB3 protein expression and promote apoptosis in gastric cancer cells.
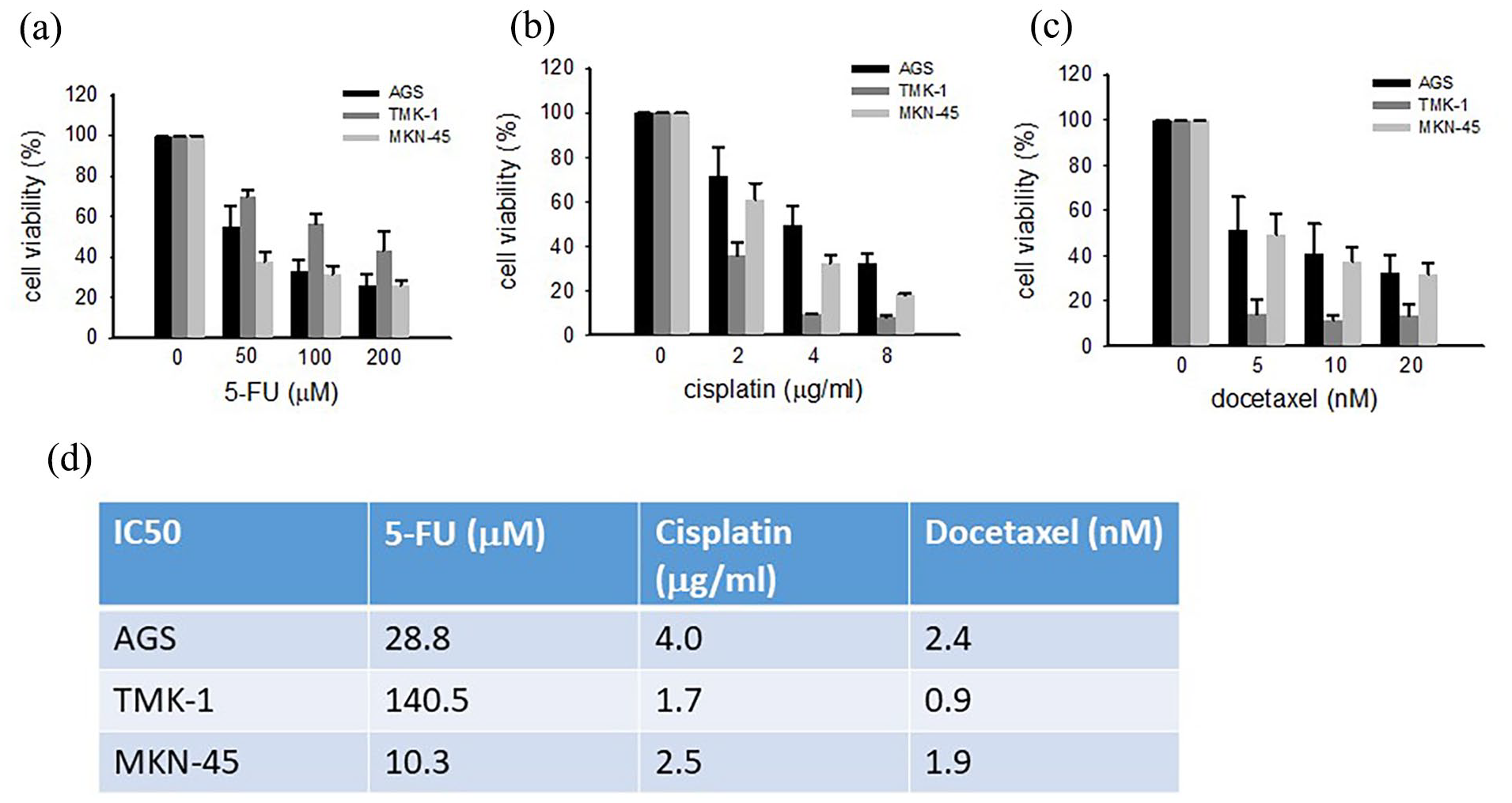
The IC50 values of 5-FU, cisplatin, and docetaxel in AGS, TMK-1, and MKN-45 cells. The cells were treated with different doses of 5-FU (a), cisplatin (b), or docetaxel (c) for 48 h, cell viability was determined by CCK8 assay (a–c), and IC50s were calculated (d). Data are representative of three to five independent experiments.

5-FU, cisplatin, and docetaxel downregulate TRIB3 and induce apoptosis. (a) AGS, (b) TMK-1, and (c) MKN-45 cells were treated with 0.5 IC50, 1 IC50, or 2 IC50 of 5-FU, cisplatin, or docetaxel for 48 h, cell extracts were prepared, and the protein levels of TRIB3 and apoptosis markers were evaluated by Western blot analysis. Data are representative of three to five independent experiments.

5-FU, cisplatin, and docetaxel increase the sub-G1 cell fraction. (a) AGS, TMK-1, and MKN-45 cells were treated with 1 IC50 of 5-FU, cisplatin, or docetaxel for 48 h. The cells were harvested and flow cytometry was used to evaluate sub-G1 cells. (b) The fractions of sub-G1 cells in AGS, TMK-1, and MKN-45 cells were calculated. Data are representative of three to five independent experiments.
Induction of autophagy by anticancer drugs
Many chemotherapeutic agents have been suggested to elicit autophagy in cancer cells.23,24 To systematically analyze this possible cellular response, we treated the gastric cancer cell lines with 5-FU, cisplatin, and docetaxel for 48 h and assessed the autophagy markers, LC3-II and p62. Western blot analysis showed that 5-FU, cisplatin, and docetaxel dose-dependently increased the levels of p62 and LC3-II in all three cell lines (Figure 4(a)–(c)). We then used the fluorescent dye, acridine orange, to stain acidic vesicular organelles autolysosomes, which are characteristic of autophagy, and found that autolysosomes were increased to varying extents in AGS, TMK-1, and MKN-45 cells treated with the tested 1 IC50 of anticancer agents (Figure 4(d)). These results suggest that 5-FU, cisplatin, and docetaxel have a certain capacity to elicit autophagy in the tested gastric cancer cell lines.

5-FU, cisplatin, and docetaxel enhance autophagy in AGS, TMK-1, and MKN-45 cells. (a) AGS, (b) TMK-1, and (c) MKN-45 cells were treated with 0.5 IC50, 1 IC50, or 2 IC50 of 5-FU, cisplatin, or docetaxel for 48 h, cell extracts were prepared, and the protein levels of p62 and LC3-II were evaluated by Western blot analysis. (d) AGS, TMK-1, and MKN-45 cells were treated with 1 IC50 of 5-FU, cisplatin, or docetaxel for 48 h. The cells were harvested and stained with acridine orange, and the amounts of acid vesicles were evaluated by flow cytometry. Data are representative of three to five independent experiments.
Overexpression of TRIB3 protects MKN-45 gastric cancer cells from ER stress-induced apoptosis
We previously reported that TRIB3 overexpression could protect gastric cancer cells against doxorubicin-induced apoptosis. 15 Here, we tested how Myc-tagged TRIB3 overexpression in MKN-45 cells impacted their response to a 48-h treatment with 5-FU, cisplatin, and docetaxel. As shown in Figure 5, TRIB3 overexpression significantly decreased the abilities of 5-FU and docetaxel, but not cisplatin, to induce PARP cleavage and caspase 3 activation. The three anticancer drug-induced upregulation of Bax was also attenuated by TRIB3 overexpression (Figure 5). Previous in vitro and in vivo studies revealed that breast, prostate, brain, pancreas, lung, and skin cancers rely on the constitutive upregulation of autophagy for their growth. 25 We thus analyzed whether autophagy modulation was involved in the ability of TRIB3 to protect gastric cancer cells from anticancer drug treatment. However, TRIB3 overexpression did not influence the induction of autophagy by anticancer drugs, as indicated by the level of LC3 II in MKN-45 cells (Figure 5). Interestingly, the overexpression of TRIB3 attenuated the anticancer drug-induced enhancements of two important regulators of ER stress, phospho-PERK and Bim (Figure 5). Together, our results indicate that attenuation of TRIB3 increases the sensitivity of gastric cancer cells to anticancer drugs, while its overexpression protects these cells from the PERK-dependent ER stress-mediated apoptotic cell death triggered by anticancer drugs.

Overexpression of TRIB3 attenuates anticancer drug-induced apoptosis. MKN-45 cells were transfected overnight with a vector encoding Myc-tagged TRIB3 and then treated with 1 IC50 of 5-FU, cisplatin, or docetaxel for 48 h. Cell extracts were prepared and the protein levels of apoptosis, autophagy, and ER stress markers were evaluated by Western blot analysis. Data are representative of three to five independent experiments.
Knockdown of TRIB3 enhances sensitivity to paroxetine
Another drug, the antidepressant paroxetine, was recently repurposed to effectively inhibit cell growth and induce apoptosis in different cancer cells. We previously showed that paroxetine exerts anticancer effects in AGS cells by inhibiting DNA repair proteins and thereby facilitating the accumulation of DNA damage, leading to apoptotic cell death. 24 To investigate whether TRIB3 could also protect gastric cancer cells against this repurposed anticancer drug, we overexpressed TRIB3 in MKN-45 cells and then applied paroxetine for 48 h. Indeed, TRIB3 proteins were dose-dependently decreased in paroxetine-treated MKN-45 cells (Figure 6(a)), and overexpression of TRIB3 rescued the paroxetine-induced cleavages of PARP and caspase 3 (Figure 6(b)). Interestingly, the overexpression of TRIB3 also attenuated the paroxetine-induced enhancement of phosphor-eIF1a, which is a downstream effector of phospho-PERK. Taken together, downregulation of TRIB3 also increases the sensitivity of gastric cancer cells to paroxetine, and overexpression of TRIB3 protects these cells from the PERK-dependent ER stress-mediated apoptotic cell death triggered by paroxetine.

Paroxetine downregulates TRIB3 and induces apoptosis in MKN-45 cells. (a) Cells were treated with different doses of paroxetine for 48 h, cell extracts were prepared, and Western blot analysis was used to determine the levels of TRIB3. (b) Cells were transfected overnight with a vector encoding Myc-tagged TRIB3 and then treated with 1 IC50 of paroxetine for another 48 h. The protein levels of apoptosis, autophagy, and ER stress markers were evaluated by Western blot analysis. Data are representative of three to five independent experiments.
In summary, our previous and present results indicate that TRIB3 can protect gastric cancer cells against anticancer drug treatment, and that downregulation of TRIB3 may increase the sensitivity of these cells to anticancer drugs. We thus suggest that the capability of anticancer drugs to downregulate TRIB3 can indicate the potential susceptibility of tumors to these anticancer drugs.
Discussion
The search for prognostic/predictive biomarkers for the responsiveness of cancer to chemotherapy is an area of active investigation that is moving from clinical features to biological determinants. The actions and regulatory mechanisms of TRIB3 are relatively well understood, but far less is known about its role in the response of cancer cells to chemotherapy. Understanding the role of TRIB3 in cellular chemotherapy responsiveness and survival could facilitate its development as a prognostic marker that could be used to improve chemotherapeutic efficiency against specific tumors. Here, we set out to evaluate whether TRIB3 downregulation could be a predictive marker for the ability of gastric cancer cell lines to respond to four chemotherapeutic agents with different DNA damage-inducing mechanisms. Indeed, we found that TRIB3 expression was negatively correlated with gastric cancer cell sensitivity to anticancer drugs, highlighting the potential of TRIB3 as a predictive biomarker. Treatment of gastric cancer cell lines with doxorubicin, 15 5-FU, docetaxel, and paroxetine dose-dependently downregulated TRIB3 expression and was associated with better overall cell survival compared to untreated cells.
As we know, distinct types of cancers display varying degrees of resistance to chemo, radio, and hormonal therapies. Drug resistances account for nearly 90% of chemotherapy fails in human cancer patients. Researchers recently explored new strategies to overcome these limitations to develop efficient, readily affordable, and safe anticancer achievements. Natural compounds, such as flavanol, terpenoids, phenolics, and alkaloids, have been reported to impact cancer and become one of the most potential strategies for cancer drug development.26,27 For example, Berberine, a benzyl tetra isoquinoline alkaloid, is effectively cytotoxic against several cancers, such as breast, colon, pancreatic, gastric, liver, oral, bone, glioblastoma, skin, and prostate cancers. Mechanically, berberine inhibits several growth factors signaling pathways, leading to inhibit cell proliferation, cell cycle progression, cell migration/invasion, and trigger apoptosis without affecting the viability of normal human cells. 26 In silico approaches, such as molecular docking and molecular dynamics simulations, have become highly effective strategies in modern drug development. Akash et al. 28 have successfully applied these strategies to identify promising Apigenin derivatives as potential inhibitors of DNA polymerase theta and treatment for human papillomavirus-associated cervical cancer. 28 In addressing multidrug resistance (MDR), several laboratory methods have been employed to evaluate drug resistance in cancer cells. These include genomic analysis of MDR tumors, drug susceptibility testing, drug influx and efflux assays, cell viability assays, and high-content and high-throughput screening techniques. 29
Our results, together with previous studies, show that TRIB3 regulates distinct cell signaling processes to influence the prognosis of diverse human cancers. As a result, it has been suggested that TRIB3 is a useful biomarker for cancer diagnosis, prognosis, prediction, and clinical strategy. TRIB3 was reported to inhibit AKT signaling and thereby suppress cancer initiation and progression. 16 In gastric cancer, high-level expression of TRIB3 is reportedly correlated with VEGF-A expression and angiogenesis, highlighting the potential of TRIB3 as a predictive biomarker for the efficacy of anti-angiogenesis treatment in gastric cancer. 30 TRIB3 plays an important role in mediating acute promyelocytic leukemia tumorigenesis and treatment response via its ability to stabilize the driver oncogene, promyelocytic leukemia-retinoic acid receptor α (PML-RARa). 31 The expression level of TRIB3 can be used to predict the responsiveness of acute promyelocytic leukemia to therapy. Moreover, a peptide compound targeting the interaction of TRIB3 with PML-RARa was shown to exert therapeutic effects and enhance such effects by other agents against acute promyelocytic leukemia. 31 Adding to these previous findings, we herein show that TRIB3 can act as a biomarker to predict the responsiveness of gastric cancer cells to DNA-damage-inducing anticancer agents, such as 5-FU, cisplatin, docetaxel, doxorubicin, and paroxetine. Our comprehensive analysis revealed that the central role of TRIB3 in ER stress and tumor progression makes TRIB3 representing not only a potential biomarker for the outcome of chemotherapy but also a potential therapeutic target for reverse drug resistance. This is consistent with previous reports that TRIB3 downregulation may enhance the efficacy of chemotherapy. For example, Hua et al. 32 demonstrated that TRIB3 increases stemness features of colorectal cancer stem cells and tumorigenesis by interacting with β-catenin and TCF4. Yu et al. 33 showed that disturbing the TRIB3-AKT interaction can accelerate Forkhead Box O1 (FOXO1) degradation and reduce SRY-Box transcription factor (SOX2) expression to suppress breast cancer stemness in mouse models of breast cancer. Therefore, manipulation of cellular TRIB3 expression may be a potentially useful strategy to cope with drug resistance.
Based on our findings and others, it is reasonable to hypothesize that if a particular drug downregulates TRIB3 in cancer cells, these cells may be more sensitive to that specific anticancer drug. Therefore, the capacity of anticancer drugs to downregulate TRIB3 could be a valuable marker for predicting tumor responsiveness, providing a useful strategy for selecting effective anticancer drugs. Moreover, manipulating cellular TRIB3 expression could be a viable strategy to enhance the effectiveness of anticancer drugs to overcome drug-resistant cancers. Alternatively, we hypothesized that TRIB3 might be a predictor to monitor the susceptibility of cancer to anticancer drugs.
Conclusion
Our results indicate that attenuation of TRIB3 increases the sensitivity of gastric cancer cells to anticancer drugs, while its overexpression protects these cells from the protein kinase RNA-like ER kinase (PERK) dependent ER stress-mediated apoptotic cell death triggered by anticancer drugs. Taken together, our previous and present results indicate that TRIB3 can protect gastric cancer cells against anticancer drug treatment and that downregulating TRIB3 may increase the sensitivity of these cells to anticancer drugs. We thus suggest that the capability of anticancer drugs to downregulate TRIB3 can indicate the potential susceptibility of tumors to these anticancer drugs.
Footnotes
Acknowledgements
We thank Dr Chun-Ying Wu (Taipei Veterans General Hospital, Taipei, Taiwan) for providing us with the AGS and TMK-1 cell lines.
Author contributions
B-HL performed TRIB3 overexpression and immunoblotting analysis of TMK-1; C-JH and Y-CH carried out immunoblotting and flow cytometry analysis of AGS and MKN-45; T-MY and S-MC conceived the study, participated in designing and coordinating the study, and contributed to writing, reviewing, and/or revising the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This paper received funding from the Research Project of Ministry of Health and Welfare, Taiwan (grant numbers 10719 and 10812).
Ethics approval
Not applicable.
Informed consent
Not applicable.
Trial registration
Not applicable.