
Abstract
Background:
The emergence and spread of antimalarial drug resistance have created an urgent need for novel therapeutic agents. Imidazolopiperazines (IZPs) represent a promising new class of compounds with broad activity against multiple stages of the malaria parasite life cycle.
Objective:
This scoping review aimed to map current evidence on the efficacy, mechanisms of action, resistance determinants, and clinical development of IZPs, particularly KAF156 (Ganaplacide) and its analog GNF179, to assess their potential as next-generation antimalarial agents.
Eligibility criteria:
Peer-reviewed studies of clinical, in vivo, or in vitro were included if they investigated IZPs for antimalarial efficacy, mechanisms of action or resistance, safety, or comparison with other drugs. Only English-language publications were considered.
Sources of evidence:
Searches were performed in PubMed, Google Scholar, ClinicalTrials.gov, and organizational databases (MMV and Novartis). Citation mapping using Litmaps identified additional related studies. The final search was completed on December 20, 2024.
Charting methods:
Two reviewers independently screened, selected, and extracted data using a form capturing study characteristics, compounds, parasite stages, efficacy, mechanisms of action or resistance, and safety. Discrepancies were resolved by consensus. Data were synthesized descriptively and summarized by study type, parasite stage, compound, and outcome.
Results:
IZPs demonstrate potent activity against asexual blood stages, liver stages, and gametocytes. Proposed mechanisms of action include mitochondrial disruption, sodium pump inhibition, and interference with protein trafficking. Resistance-associated mutations have been identified in genes such as Pfugt, Pfact, and Pfcarl. Clinical trials of KAF156, including combination therapy with lumefantrine, report favorable efficacy, safety, and tolerability, with side effects that are generally mild and manageable.
Conclusion:
IZPs hold strong potential as next-generation antimalarial agents, particularly for combination therapies. Future research should prioritize clarifying mechanisms of action, monitoring resistance pathways, and optimizing clinical deployment strategies to address the ongoing challenges of antimalarial drug resistance and support malaria elimination goals.
Introduction
Recent evidence suggests that Plasmodium falciparum partial resistance to artemisinin has been well characterized in Southeast Asia1–4 and also confirmed in several African countries.5–7
In sub-Saharan Africa, artemisinin derivatives and artemisinin combination therapies (ACTs) have been the pillar of malaria treatment for over a decade. 8 By the mid-2010s, the World Health Organization (WHO) reported that most African countries had adopted ACTs as the first-line therapies. 9 In practice, ACTs remain widely used and highly effective across the continent. 10 Indeed, more than 2 billion ACT courses were delivered globally between 2010 and 2023, 11 reflecting the immense drug pressure P. falciparum is subjected to in Africa.
Widespread ACT treatment failure has not yet been observed in Africa. WHO notes that no resistance to ACT partner drugs has been confirmed on the continent and that overall ACT efficacy remains high. 10 Currently, evidence shows that artemisinin resistance is emerging in sub-Saharan Africa, with several validated and candidate Pfk13 mutations now reported across the continent. The R561H mutation, first identified in Rwanda in 2014, 6 has reached a prevalence of 20%–22% in some regions and has been associated with delayed parasite clearance. 12 Other important resistance-associated mutations include M476I in Tanzania, 13 A675V in Uganda, 14 Nigeria, Kenya, 13 P574L in Rwanda 6 and Uganda, 14 C469Y in Uganda, R622I in Ethiopia, 15 P553L in Ghana 16 and Angola 13 , F446I in Mali, 13 and C580Y in Ghana. 17
Therefore, the spread of artemisinin resistance in Africa remains a significant concern due to the potentially devastating consequences.5,18 The WHO has launched a new strategy to address emerging antimalarial drug resistance in Africa, emphasizing the importance of surveillance, optimizing diagnostics and therapeutics, and limiting the spread of resistant parasites. 19 This strategy aims to minimize the threat and impact of antimalarial drug resistance in Africa, where the burden of malaria is highest globally. 20 Despite the challenges posed by resistance, ACTs remain the recommended treatment for uncomplicated P. falciparum malaria. Adequate surveillance and the development of new antimalarial candidates like imidazolopiperazines (IZPs), OZONIDE, and Plasmodium phosphatidylinositol 4-kinase (PI4K) inhibitors, among others, will be crucial in the battle against malaria drug resistance. 21 Promising candidates include IZPs KAF156 (Ganaplacide) and its close analog GNF179, which are effective against Plasmodium asexual blood-stage infections, active against the liver stage parasite, prevent transmission, and block infection in animal models. In preclinical and clinical studies, IZP compounds, KAF156 and GNF179, have demonstrated significant antimalarial efficacy, making them valuable candidates for malaria treatment and the elimination agenda.22–24 Ongoing clinical evaluations assessing KAF156 safety and efficacy in malaria treatment and the understanding of IZPs’ mechanisms of action will offer a novel approach to malaria treatment that could overcome existing drug resistance mechanisms.
Rationale
Given the increasing threat of artemisinin resistance in Africa and the urgent need for novel antimalarial therapies, a scoping review is needed to map the existing evidence on IZPs as an advanced class of antimalarial candidates. This review is expected to provide a comprehensive synthesis of their efficacy, safety profiles, mechanisms of action, resistance determinants, and potential role in future malaria control strategies.
Objectives
This scoping review aims to (i) summarize and synthesize the available evidence on the efficacy of IZPs as antimalarial drug candidates from past studies; (ii) clarify the mechanisms of action and resistance proposed across different studies; and (iii) discuss the current body of literature to inform future IZP development strategies.
Methods
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) was used in the design, conduct, and report of this scoping review (Supplemental File 1).
Protocol and registration
The protocol for this scoping review was developed in accordance with the PRISMA-P guidelines and refined by the review team. The protocol has not been submitted to any online registry, but is provided as Supplemental File 2.
Eligibility criteria
Studies were eligible for inclusion if they were published in peer-reviewed journals and focused on one or more of the following aspects of IZPs: laboratory, preclinical, or clinical efficacy; mechanisms of action; mechanisms of resistance; comparative analysis with other antimalarials; safety profiles. Inclusion criteria included studies of any design (randomized controlled trials, observational clinical studies, in vitro experiments, and in vivo animal studies) that directly addressed these outcomes. Only studies published in English were included, and no geographic restrictions were applied. Studies were excluded if they were not focused on IZPs or did not report relevant data on efficacy, mechanisms of action, or resistance; if they lacked original data, such as commentaries; if they were duplicate publications identified during the search; or if they were conference abstracts without sufficient methodological or results detail.
Information sources
To identify relevant studies, searches were conducted across multiple electronic databases and resources, including clinical trial registries (mainly ClinicalTrials.gov and the International Clinical Trials Registry Platform), bibliographic databases (PubMed and Google Scholar), and organizational resources such as the Medicines for Malaria Venture (MMV) and Novartis websites. Litmaps was used to establish the link between published articles on IZPs and other topics of interest related to this review. The most recent search was performed on December 20, 2024.
Search strategy
Searches combined the following terms using Boolean operators (AND/OR): “Imidazolopiperazines,” “IPZ,” “IZP,” “antimalarial,” “antimalarial candidates,” “Plasmodium,” “Plasmodium falciparum,” “Plasmodium berghei,” “Plasmodium vivax,” “KAF156,” “GNF179,” “malaria treatment,” “drug resistance,” “artemisinin-resistant,” “clinical trials,” and “antimalarial efficacy.” Searches were limited to English-language publications up to the date of December 20, 2024.
Selection of sources of evidence
Two reviewers independently screened all titles and abstracts identified by the search strategy. Full texts were obtained for studies that met the inclusion criteria or where eligibility was unclear. Discrepancies between reviewers were resolved by discussion and consensus with the other co-authors from our research team.
Data charting process
Data from included studies were extracted independently by two reviewers using a standardized data-charting form. The form was developed iteratively to capture key study characteristics and outcomes, including the compound tested, study design, parasite stage, efficacy results, mechanisms of action, resistance markers, and safety data. Any disagreements in data extraction were resolved through discussion between the authors.
Data items
The variables extracted from each study included study characteristics (year, country, study design, sample size), intervention characteristics (IZP compound, dosage, duration), outcomes (preclinical and clinical efficacy, safety, tolerability, pharmacokinetics), and mechanisms of action and resistance (gene mutations, cellular pathways, pharmacologic targets). Comparative data with other antimalarials were also extracted when available.
Critical appraisal of individual sources of evidence
No formal risk of bias or quality assessment was performed, given the heterogeneity of study designs and outcomes. Instead, a descriptive synthesis was conducted on IZPs to highlight methodological strengths, current evidence, gaps in the literature, and future directions to consider.
Synthesis of results
The charted data were summarized descriptively. Studies were grouped by parasite stage targeted (asexual blood stage, liver stage, gametocytes), type of study (in vitro, in vivo, clinical trials), IZP compound (KAF156, GNF179), and outcomes assessed (efficacy, safety, mechanism, resistance). Findings were presented in figures, tables, and narrative synthesis, emphasizing patterns in efficacy, resistance mechanisms, and safety profiles, while identifying research gaps and directions for future investigation.
Results
Selection of sources of evidence
A total of 62 studies were identified through database searches. After removing seven duplicates, 55 records were screened based on titles and abstracts. Of these, 23 studies were excluded for not meeting the eligibility criteria, leaving 32 articles for full-text review. Another set of 09 studies was removed for not focusing on IZPs. In total, 23 studies were included in the final synthesis. The selection process and reasons for exclusion at each stage are summarized in Figure 1.
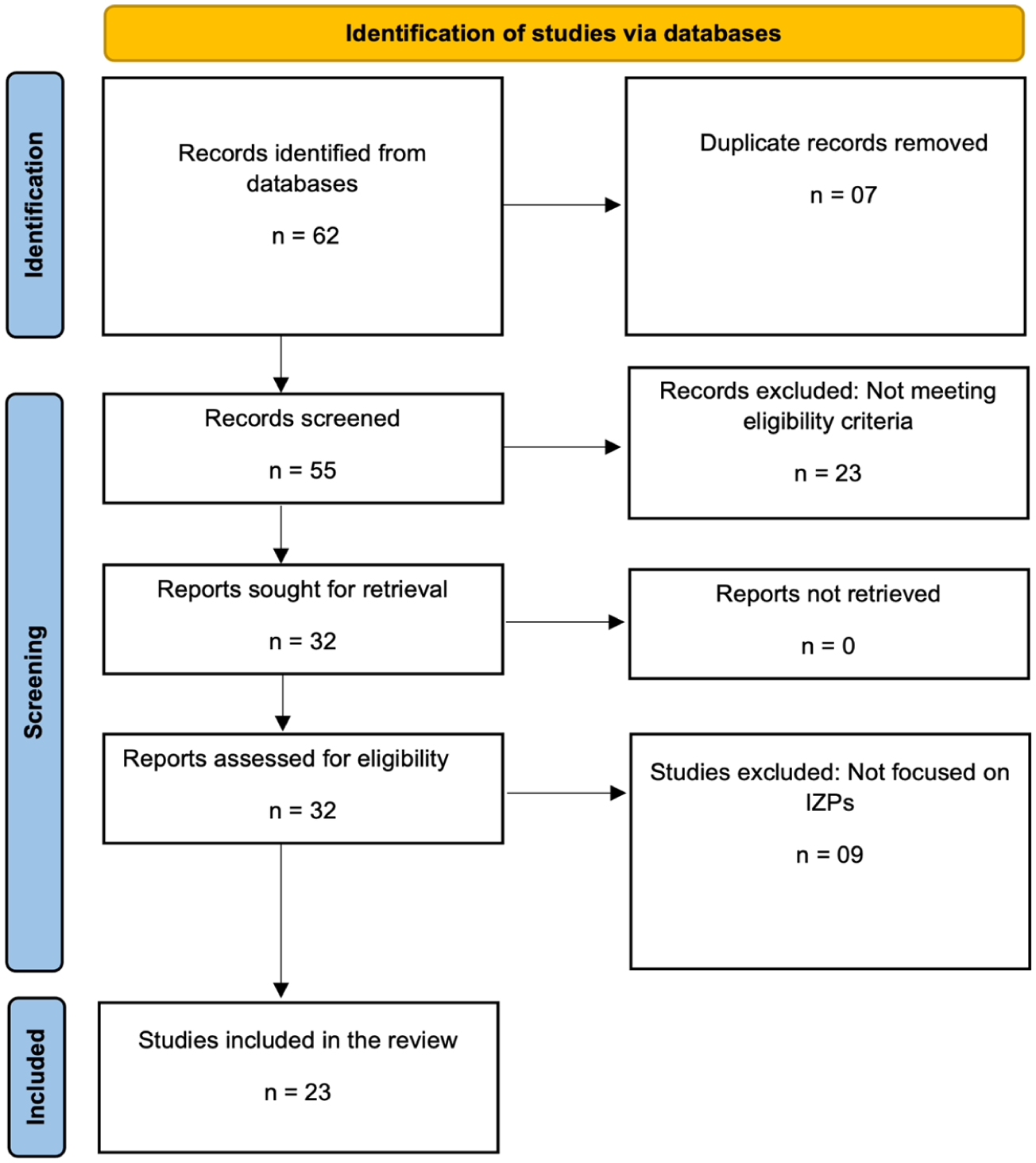
Flow chart of literature search on imidazolopiperazines according to the PRISMA-ScR.
Characteristics of sources of evidence
The included studies encompassed in vitro laboratory investigations, in vivo animal studies, and clinical trials. These studies were conducted across diverse geographic regions and investigated two primary IZP compounds: Ganaplacide and its analog GNF179. Key study characteristics, such as study design, parasite species tested, sample sizes (for human-based trials), dosage regimens, outcomes evaluated, and key findings, are presented in Table 1.
Summary of studies on key efficacy and safety data for KAF156 and GNF179.
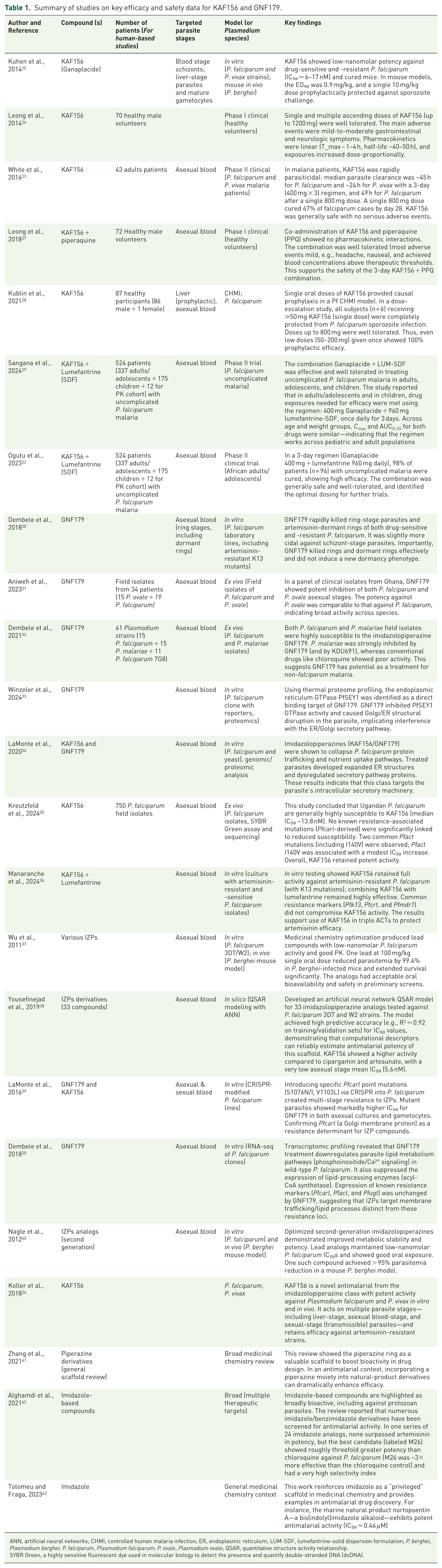
ANN, artificial neural networks; CHMI, controlled human malaria infection; ER, endoplasmic reticulum; LUM-SDF, lumefantrine-solid dispersion formulation; P. berghei, Plasmodium berghei; P. falciparum, Plasmodium falciparum; P. ovale, Plasmodium ovale; QSAR, quantitative structure activity relationship.
SYBR Green, a highly sensitive fluorescent dye used in molecular biology to detect the presence and quantify double-stranded DNA (dsDNA).
Results of individual sources of evidence
Preclinical studies assessed parasite stage specificity, potency against resistant strains, and potential mechanisms of action. In vivo animal studies reported on pharmacokinetics, efficacy across different malaria models, and safety profiles. Clinical trials evaluated the therapeutic efficacy, safety, and tolerability of KAF156 in monotherapy and in combination with partner drugs. Resistance studies provided insights into genetic mutations and molecular pathways associated with reduced susceptibility. A detailed list of studies included in the narrative is presented in Table 1, which summarizes the main outcomes and relevance to the objectives of the current review.
Synthesis of results
Discovery and development of IZPs as an effective treatment for malaria
Chemical structure and properties of IZPs
IZP compounds are members of a larger class of heterocyclic amines characterized by a fused ring structure consisting of a piperazine ring and an imidazole ring 37 (Figure 2). The imidazole ring, with five members and two nitrogen atoms, is known for its involvement in numerous biological processes, accounting for some of the pharmacological effects of IZP compounds. The piperazine ring increases membrane permeability, which helps the drug migrate to the targeted sites in the body. 41 The compounds’ stability and bioavailability are enhanced by their structural basis, which gives them appealing possibilities for drug development. 44

General structure of IZP 37 and substituent variations leading to KAF156 and GNF179 compounds.
Early GNF hits and first-generation leads showed strong cellular potency but suffered from metabolic liabilities and limited oral exposure in vivo; as a result, the series underwent iterative SAR (Structure–Activity Relationship) and core modifications to improve microsomal stability and mouse oral exposure. Second-generation changes (including 8,8-dimethyl substitutions on the piperazine core and the para-halogenation at R₂/R₃) substantially enhanced metabolic stability and oral bioavailability, leading to the preclinical/clinical candidates KAF156 and GNF179 with superior pharmacokinetic profiles and efficacy in animal models. 37
The IZP scaffold consists of three modifiable positions (R₁, R₂, and R₃) that were optimized through medicinal chemistry to improve potency, stability, and safety (Figure 2). At R₁, the acyl-amino side chain is typically a glycine-derived acetamide (–CO–CH₂–NH₂), where the free amino group is essential for activity; the addition of an α-methyl substituent enhances metabolic stability (higher C-max and are under the curve
Early exploration and versatility of IZPs
IZPs are an adaptable class of chemicals with a broad spectrum of biological activity.30,31,34,46,47 Early medicinal chemistry programs considered IZP scaffolds for broader therapeutic applications, given their favorable physicochemical properties such as blood–brain barrier penetration. 42 Their selective cytotoxic effects against specific cancer cell lines led to their eventual expansion into other fields, such as oncology. This versatility of IZPs highlights their potential for broader therapeutic applications beyond malaria and oncology, emphasizing their significance in drug development across various fields. Medicinal chemistry has explored IZP for its versatility and effectiveness across a range of therapeutic domains. 43 These compounds have been explored in cancer treatment and have shown efficacy in specific cancer cell lines like breast, colon, and pancreatic cancer cells. 42 While not advanced clinically in these areas, related derivatives have shown activity in oncology and infectious disease models. 42
In the early 2000s, IZPs were identified through chemical screening programs aimed at discovering novel pharmacological scaffolds (Figure 3). As described above, the piperazine core with its three modifiable sites offered flexibility for optimization, making IZPs attractive starting points for drug discovery. 40

Evolution of research on IZPs from discovery to development.
A new global agenda toward malaria elimination and eradication has highlighted the need for novel antimalarial agents with activity against multiple stages of the parasite life cycle and diverse parasite species. The development phase (the early 2010s) of IZP research has produced a promising molecule, KAF156, and its close analog, GNF179. IZP (KAF156 and GNF179) meets the new global agenda malaria eradication criteria for novel antimalarial agents, as it has demonstrated excellent efficacy and safety in phase IIb clinical trials against P. falciparum and P. vivax species23,25,31,46,47 while displaying potent inhibition activity against P. malariae and P. ovale.23,25,31,46,47 IZPs have shown malaria transmission-blocking properties in vitro and in vivo.23,25,31,46,47 They were also proven to be effective against both liver46,48 and blood stages parasites25,26,45,46 (Figure 4). IZP compounds have since been extensively studied for their pharmacological properties toward approval as a malaria treatment.

Key genes impacted by GNF179 (100 nM) treatment in P. falciparum Dd2 wild-type and its mutant parasites resistant to IZP. 49
These targeted substitutions yielded the clinically advanced compounds KAF156 (Ganaplacide) and GNF179, both of which exhibit improved potency, metabolic stability, and oral bioavailability compared to earlier analogs such as GNF707, GNF452, 37 and GNF943, which displayed promising activity but limited pharmacokinetic profiles (Figure 3). 40 While these earlier compounds showed low-nanomolar potency against P. falciparum (EC₅₀ ≈ 3–15 nM), they suffered from poor metabolic stability, rapid clearance, and limited oral exposure, preventing further development.37,40
The exploration of KAF156 and GNF179 as antimalarial agents has been of great interest in recent pharmaceutical research, which included in vitro and in vivo tests, and human clinical trials. They have covered aspects like in vitro potency and drug mechanism of action and resistance, drug in vivo efficacy, safety, and the potential of these IZPs in treating human malaria.
Efficacy and safety of IZPs
In vitro studies have demonstrated that IZPs, particularly KAF156 and GNF179, are highly potent against multiple Plasmodium species, including P. falciparum, 46 P. ovale, 31 P. malariae, 32 P. berghei in vitro, 50 P. vivax,23,25 even in strains resistant to existing antimalarial therapies.30,36 An ex vivo study conducted on up to 750 P. falciparum field isolates concluded that all the strains were highly susceptible to KAF156 (median IC₅₀ ~ 13.8 nM). 35 This study reported two common Pfact mutations (including I140V) as associated with a modest IC₅₀ increase. 35 Ganaplacide exhibited a higher potency compared to cipargamin and artesunate in a computational model assessing the effect of drugs on various parasite stages. On the asexual stage, KAF156 showed a mean IC50 of 5.6 nM, slightly higher for male gametocytes (6.9 nM) and 47.5 nM for the female gametocytes. 51
These IZP compounds exhibited broad-spectrum activity by targeting asexual blood stages, liver stages,25,46 and gametocytes, 25 showing their potential to block transmission46,47 as well as treat acute infection. However, gametocytes aside, IZPs have not yet been evaluated against mosquito stages such as ookinetes, oocysts, or sporozoites, which represents a gap for future transmission-blocking studies. In vitro assays further suggest that KAF156 and GNF179 may interfere with mitochondrial functions and sodium homeostasis, impairing parasite growth. 30 Other in vitro studies confirmed that KAF156, both alone 51 and in combination with lumefantrine, 36 was effective against P. falciparum strains carrying clinically validated artemisinin-resistant kelch13 mutations (R539T, C580Y, G449A), 51 including those with mutations in Pfk13 (drug-pressured M476I, gene-edited R561H 3D7, SEA-field isolates R539T and C580Y) and in a quiescent state. 36 The artemisinin-resistant strains also carried mutations on Pfcrt (M74I, N75E, and K76T), Pfmdr1 (N86Y and Y184F), Pfmdr2 (F423Y), Pfdhps (437G, K540N, and A581G), and Pfdhfr (N51I, C59R, and S108N) genes. 36 The study concluded that KAF156, both as a monotherapy and in combination with lumefantrine, was effective against artemisinin-resistant parasites, including strains in a quiescent state. 36
Thus, IZPs were well-tolerated at therapeutic doses with no significant adverse effects observed. 27 KAF156, administered at doses up to 15 mg/kg, 46 showed no major hematological, renal, or hepatic toxicity.27,28 In vitro studies in human cell lines further confirmed the absence of significant cytotoxicity at relevant concentrations.50,52 Notably, in animal models, KAF156 exhibited potent activity against both the blood and liver stages of Plasmodium spp., significantly reducing parasitaemia in recurrent infection models, with no major hematological, renal, or hepatic toxicity observed at therapeutic doses.25,37,40
Clinical efficacy of IZPs (on humans)
Phase I clinical trials primarily focused on assessing the safety, tolerability, and pharmacokinetics of KAF156 and GNF179 in healthy human volunteers. Common adverse events reported in a few studies were minor and included sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.26–29 KAF156 was well-tolerated at doses up to 800 mg in one phase II study involving 70 healthy male participants, 23 and up to 1200 mg in a separate phase II trial. 26 The most commonly reported adverse events were mild and self-limiting gastrointestinal and neurological effects, such as nausea, headache, and asymptomatic bradycardia. 26 No participants were withdrawn from this study due to adverse events. Further phase I analysis of pharmacokinetics indicated that KAF156 has a predictable absorption profile, with no significant interactions with food or other medications. The study also found no major safety concerns related to hematological, renal, or hepatic toxicity. 27
The phase I evaluation of the pharmacokinetic/pharmacodynamic interaction of KAF156 and piperaquine showed that the co-administration of piperaquine and KAF156 led to an increase in the maximum plasma concentration values of the two molecules. 27 Early clinical trials have focused on assessing the safety, tolerability, and pharmacokinetics of KAF156 in healthy volunteers and individuals with uncomplicated P. falciparum or P. vivax malaria. 26 Other trials exploring the effectiveness of KAF156 showed the potential for rapid clearance of parasites and prevention of disease recurrence. 23
KAF156 went through efficacy evaluation in treating uncomplicated malaria caused by P. falciparum and P. vivax in phase II trials. A multicenter phase II study conducted in Thailand and Vietnam assessed the efficacy of two dosing regimens: a 3-day 400 mg daily dose and a single 800 mg dose. 23 Among the 21 patients receiving the single 800 mg dose, 67% achieved parasite clearance within 48 h with no serious adverse effects. 23 KAF156 exhibits dose-dependent efficacy, with complete protection achieved at doses between 100 and 800 mg. Partial protection was also observed at 50 mg (21.4% efficacy), while no protection was noted at 20 mg. 28
Ongoing phase IIb trials are evaluating the safety and efficacy of KAF156 in combination with lumefantrine. KAF-lumefantrine combination therapy, like previous ACTs, aims to improve treatment outcomes and reduce the risk of resistance development. In a randomized phase II trial of 524 patients, the oral KAF156–lumefantrine solid dispersion formulation achieved cure rates (~95%) comparable to artemether–lumefantrine, 53 with the added benefit of activity against artemisinin-resistant parasites.22,29 Safety profiles are also broadly similar, with the combination being generally well-tolerated and adverse events limited to mild gastrointestinal and neurological symptoms, with manageable side effects such as headache, nausea, and fatigue. 23 KAF156, used as a combination with piperaquine in a pharmacokinetic study in healthy volunteers, revealed no adverse safety or cardiac interactions. 27
In a multinational phase II study involving 446 participants (adults, adolescents, and children), KAF156 was tested in combination with lumefantrine solid dispersion formulation and showed great promise as a treatment for uncomplicated P. falciparum malaria. 29 In this study, conducted from 2017 to 2021, the treatment was tested on adults, adolescents, and children, and it proved both effective and well-tolerated. The pharmacokinetics of the combination revealed no significant interactions between the two drugs, with KAF156 exposure increasing predictably with dose. A daily dose of 400 mg of KAF156 and 960 mg lumefantrine over 3 days was enough to achieve the drug levels needed for effective treatment against P. falciparum. This regimen worked well across different age and weight groups. 29 This study opened the path to various formulations of KAF156-based combinations to improve malaria treatment options across varied patient populations. Ongoing research is examining KAF156 in single-dose use25,26,28 and in combination with other drugs, such as dihydroartemisinin, pyronaridine, piperaquine, monomethyl-amodiaquine, and lumefantrine, KAF156, to increase efficacy.22,27,29,36
IZPs, currently in phase IIb clinical trials, demonstrated efficacy in patients with P. falciparum and P. vivax malaria.23,54 They showed promise in treating symptomatic asexual blood-stage infections, preventing transmission, and blocking infection in humans34,45,55 and animal models. 25 KAF156 has been proven to act on multiple stages of the malaria parasite’s life cycle, both in vitro25,34 and in vivo.23,24 Current trials include ongoing phase IIb studies in Africa and Asia that aim to characterize the activity, efficacy, and toxicity of the combination of KAF156 and lumefantrine in adults, children, and infants. 56
IZP drug resistance and drug mechanism of action
Mechanism of action of IZPs
IZP compounds represent a significant advancement in the fight against malaria due to their ability to target specific biological pathways of the Plasmodium parasite, including strains resistant to current treatments. Various approaches have been used to better understand their modes of action. These studies included transcriptomic analyses 49 along with metabolic studies, functional genetics, and subcellular localization analyses. 34 IZPs represented by KAF156 and GNF179 disrupt critical cellular processes in P. falciparum, including lipid metabolism, intracellular protein trafficking, and calcium signaling, ultimately disrupting several critical cellular processes in P. falciparum.34,49 These include alterations in lipid metabolism, intracellular protein trafficking, and calcium homeostasis, which together compromise parasite survival (Figure 4). However, it is not yet established which of these represents the primary molecular target and which are downstream consequences of the drug exposure.
Transcriptomic and metabolic analyses suggest that IZPs induce a downregulation and marked inhibition of lipid metabolism and membrane transport. The downregulation of key enzymes (such as acyl-CoA synthetase, lipases, and esterases) prevents the production of diacylglycerol (DAG), a crucial lipid involved in intracellular signaling. 49 The absence of DAG directly interferes with intracellular calcium (Ca²⁺) regulation, affecting key enzymes such as protein kinase C (PKC) and calcium-dependent protein kinases (CDPK).57,58 This imbalance alters membrane permeability, disrupts lipid transport, and disorganizes parasite membranes, ultimately compromising parasite survival. 59 The impact on membrane trafficking is particularly evident in the endoplasmic reticulum (ER) and parasitophorous vacuoles, blocking the export of essential proteins required for parasite survival.60,61 As the reflection of these transcriptomic changes on their corresponding alterations in protein abundance is to be confirmed, further proteomic and biochemical validation is required.
Furthermore, genetic and imaging approaches have demonstrated that IZPs primarily target the parasite’s intracellular secretion network by blocking protein maturation and trafficking within the ER and Golgi apparatus. Experiments combining targeted mutagenesis and genome sequencing have shown that parasites exposed to IZPs develop alterations in genes involved in intracellular trafficking regulation and lipid biosynthesis. 34 According to the same study, the use of fluorescent probes revealed an accumulation of IZPs in the ER, leading to abnormal expansion of this structure and inducing ER stress, which results in the premature degradation of misfolded proteins in genetically modified Saccharomyces cerevisiae. Consequently, treated parasites fail to export essential proteins to the host erythrocyte and parasite compartments, preventing the establishment of new membrane permeabilities required for nutrient acquisition (Figures 4 and 6). This study combined work in P. falciparum and in genetically modified S. cerevisiae, making these data indicative but not definitive for the processes occurring in Plasmodium. IZP-resistant parasites exhibit slower growth than wild-type parasites in the absence of the drug, indicating a fitness cost likely associated with sub-optimal activity of the mutated gene. 28 While informative, these results should be considered indirect and complementary rather than definitive evidence in Plasmodium.
The lethal effect of IZPs thus relies on inhibiting lipid metabolism, calcium transport, and intracellular protein trafficking.34,49 These effects disrupt multiple stages of the parasite’s life cycle, including blood, liver, and gametocyte stages (Figure 5). This multifaceted approach makes IZPs a major breakthrough in the development of new antimalarial therapies.
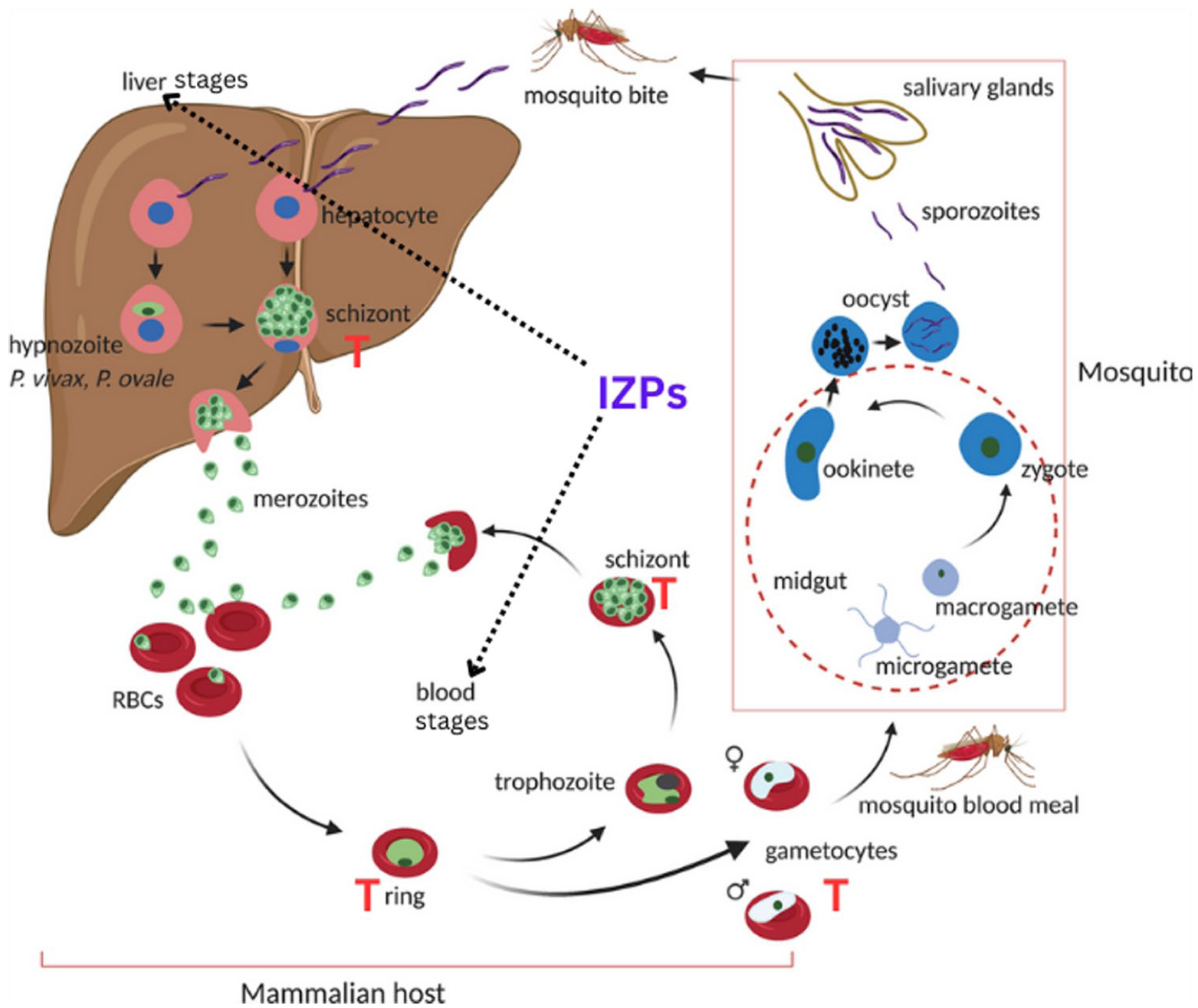
IZPs multistage mode of action in inhibiting malaria transmission and asexual parasite development.

Proposed effects of IZPs on malaria parasite P. falciparum wild-type organelles.
Despite numerous studies proposing various mechanisms of action for IZPs, their exact mode of action remains complex and unclear. To precisely understand their exact mode of action, more in-depth cellular approaches are needed. The use of high-resolution imaging and functional genetic screens could help pinpoint their subcellular targets more accurately. Furthermore, in-depth investigations into their interactions with key cellular pathways and potential off-target effects will be essential to fully elucidate their mechanism of action.
Antimalarial drug resistance
The mechanism of P. falciparum resistance to IZPs relies on a multifactorial adaptation combining genetic mutations, 34 ER and Golgi membrane transporters, along with transcriptomic reprogramming 49 affecting lipid metabolism and intracellular trafficking. Genetic analysis has revealed that mutations in three key genes, Pfcarl, Pfact, and Pfugt39,52,55 enable resistant parasites to bypass intracellular transport blockages and maintain efficient protein export despite the inhibitory effects of IZPs. Specific resistance-associated SNPs have been described, including Pfcarl L830I, S1076I, V1103L, and I1139K; Pfact Q116K, A150S, and F404I; and Pfugt V200I and F37V, which are associated with measurable phenotypes EC₅₀ fold-shifts in resistant parasites. These mutations modify ER and Golgi dynamics, reducing the accumulation of unprocessed proteins and facilitating their transport.34,49 The role of Pfact in resistance is conserved across species, since analogous mutations in Pbact of P. berghei have also been linked to IZP resistance. More specifically, mutations in Pfcarl stabilize newly synthesized proteins and facilitate their delivery to the Golgi, 39 while those in Pfact regulate the import of acetyl-CoA into the ER, an essential precursor for post-translational protein modifications. As for Pfugt, it plays a crucial role in protein glycosylation and lipid homeostasis, contributing to the protection of parasite membranes and limiting the toxicity associated with the accumulation of misfolded proteins. In addition, transcriptomic 49 analyses have revealed a reorganization of gene expression, particularly in pathways related to lipid metabolism, calcium signaling, and protein folding. These transcriptional changes may represent compensatory responses to drug pressure rather than primary resistance mechanisms, and further work is needed to clearly distinguish cause from effect. To counteract the inhibition of lipid metabolism, these parasites upregulate lipases and acyl-CoA synthetases, preserving lipid production and maintaining membrane fluidity.
Because IZP resistance can arise rapidly through genetic and metabolic adaptations like previous antimalarials, a key strategy to safeguard their clinical utility has been to evaluate them in combination with partner drugs. Therefore, to mitigate the development of resistance, various combinations involving KAF156 with other antimalarials are under investigation for their synergistic efficacy and ability to delay resistance emergence.22,27,36 These combinations may delay resistance through complementary half-lives, activity across multiple parasite stages, and reduced probability of resistant clones surviving treatment. In particular, KAF156–piperaquine and KAF156–lumefantrine combinations are being evaluated as strategies for their potential to delay the emergence of IZP-resistant strains. These studies have so far shown no evidence that resistance arises rapidly with combination formulations,22,27,29 supporting their potential role in resistance management based on their synergistic actions.
Discussion
IZPs (at this stage, KAF156 and GNF179) continue to show a compelling preclinical and early-to-mid clinical profile covering liver, asexual blood, and transmission stages, including activity against artemisinin-resistant parasites.23,25,34,46,47 Early human studies established proof-of-concept antimalarial activity and an acceptable short-term safety/tolerability profile, while phase II combination studies with lumefantrine suggest a clinically deployable path if efficacy and safety are confirmed in phase III.22,23,26,28,29,53
Multiple approaches converge on a primary perturbation of the parasite secretory/trafficking machinery, with IZPs accumulating in the ER and disrupting protein maturation and export. 34 Resistance selections have repeatedly mapped to transport and trafficking genes, Pfcarl, Pfact, and Pfugt, implicating these membrane transporters in modulating IZP susceptibility, likely by altering ER–Golgi dynamics or metabolite flux.39,52,55 These genetics-anchored findings are complemented by transcriptomic responses indicating remodeling of lipid metabolism, protein folding, and Ca²⁺ signaling under IZP pressure; however, these transcriptional shifts should be interpreted cautiously as many may be compensatory rather than causal. 49 Together, the data support a model in which IZPs target the intracellular secretory pathway, with resistance emerging through transporter-mediated adaptation.
Strengths of the current evidence base include reproducible multistage activity in vitro and in vivo, specifically, against ring and dormant-ring stages and artemisinin-resistant backgrounds,25,30,34,46,47,51 translational agreement between mechanistic hypotheses (secretory blockade) and parasite phenotypes, and clinically compatible pharmacokinetics for once-daily short courses when paired with a long-acting partner.22,26,27,29
Our review focused specifically on IZPs and did not attempt a comprehensive comparison across all novel antimalarial classes. Nonetheless, available in vitro work that assessed Ganaplacide alongside other agents (such as the PfATP4 inhibitor cipargamin) indicates that multiple mechanisms may retain activity against artemisinin-resistant parasites, though they differ in stage specificity, resistance liabilities, and combination ways.36,51
Given transporter-mediated resistance pathways and the multifaceted compensatory responses observed under IZP pressure, combination therapy remains essential. The KAF156–lumefantrine combination provides a proper example: Lumefantrine offers a long terminal half-life and a distinct hemozoin-related mechanism, potentially “covering” residual parasites after rapid IZP-driven clearance; in vitro work supports activity of the pair against artemisinin-resistant parasites, and clinical pharmacokinetic analyses to date have not identified problematic drug–drug interactions.22,29,36 Separately, a phase I PK/PD study indicates KAF156–piperaquine lacks major PK liabilities, supporting its exploration for chemoprevention or treatment where a long-tail partner is desirable. 27
Deployment choices for IZP-based combinations will intersect with evolving ACT performance and WHO guidance on resistance containment, including surveillance and multiple first-line therapy strategies.10,18–20 If phase III results are prominent, prioritizing regions with documented artemisinin partial resistance but preserved partner-drug efficacy may yield the greatest near-term impact.
Prospective molecular surveillance for Pfcarl, Pfact, and Pfugt polymorphisms (and any emergent loci from field failures) should be integrated into routine monitoring alongside clinical efficacy studies and in vitro susceptibility testing.34,39,49,52,55 Where feasible, transcriptomic or proteomic adjuncts could help distinguish pre-existing resistance from treatment-induced compensatory states in breakthrough infections. Building these elements into phase III and early implementation will accelerate detection of resistance patterns and inform timely policy updates.
Future research on IZPs should focus mechanistically on resolving the direct molecular target(s) within the secretory pathway, structurally and functionally mapping resistance-associated transporter variants, and linking transcriptomic remodeling to fitness costs and collateral sensitivities that may be exploitable in drug combinations.33,34,39,49,52 On the clinical side, phase III trials across diverse epidemiologic settings, podiatric and pregnancy studies, and refined combination dosing strategies are critical next steps. Evaluating chemoprevention and relapse-prevention potential in light of pharmacokinetic properties will also be important.22,26–29,53 At the policy level, aligning IZP deployment with WHO resistance-response strategies, integrating post-marketing surveillance, and preparing cost-effectiveness analyses will be essential to inform adoption in high-burden and low-resource settings.
Limitations
This scoping review has some limitations. First, although a systematic search strategy was employed across multiple databases, we restricted our inclusion to studies published in English. This may have led to language bias and the exclusion of potentially relevant evidence from non-English sources. Second, given the heterogeneity of study designs, ranging from in vitro and in vivo experiments to early-phase clinical trials, we did not perform a formal risk-of-bias or quality assessment. Instead, we relied on descriptive synthesis, which limits the strength of inference regarding comparative efficacy and safety.
At the evidence level, most clinical data for IZPs are derived from phase I and II trials with relatively modest sample sizes, limited follow-up durations, and restricted geographic representation. As such, the generalizability of findings to diverse endemic settings and resistant parasite backgrounds remains uncertain. Finally, because this review focused specifically on IZPs, we did not conduct a comprehensive assessment of other novel antimalarial classes, which may limit the broader contextualization of IZPs within the drug development pipeline.
Conclusion and future perspectives
IZPs possess a broad spectrum in terms of effectiveness. They are active against various life stages of Plasmodium and current drug-resistant strains. Proposed complex mechanisms of action make them excellent candidates for combination therapies with existing antimalarials, for better efficacy, reducing treatment durations and failure, and helping manage drug resistance. While initial clinical trials indicate a favorable safety profile for current IZPs used as antimalarials, ongoing phase III trials, long-term safety data, and post-marketing surveillance are needed to identify rare or delayed adverse effects. To date, IZPs are among the leading novel classes of antimalarials currently in clinical development. Future research should therefore focus on both the full elucidation of their mechanism of action and understanding how resistance-associated mutations alter parasite biology. Also, at the clinical level, further trials are needed to define optimal combination strategies, to assess efficacy across diverse endemic regions, and to extend testing to the most vulnerable populations, particularly children and pregnant women. Such studies may considerably contribute to establishing the greater impact and long-term role of IZPs in next-generation malaria treatment and control strategies.
Supplemental Material
sj-docx-1-tai-10.1177_20499361251401771 – Supplemental material for Imidazolopiperazines as next-generation antimalarial agents: a scoping review of efficacy, mechanisms of action and resistance; prospects for future development
Supplemental material, sj-docx-1-tai-10.1177_20499361251401771 for Imidazolopiperazines as next-generation antimalarial agents: a scoping review of efficacy, mechanisms of action and resistance; prospects for future development by Fatoumata Ousmane Maiga, Laurent Dembele, Mohamed Maiga and Abdoulaye A. Djimde in Therapeutic Advances in Infectious Disease
Supplemental Material
sj-docx-2-tai-10.1177_20499361251401771 – Supplemental material for Imidazolopiperazines as next-generation antimalarial agents: a scoping review of efficacy, mechanisms of action and resistance; prospects for future development
Supplemental material, sj-docx-2-tai-10.1177_20499361251401771 for Imidazolopiperazines as next-generation antimalarial agents: a scoping review of efficacy, mechanisms of action and resistance; prospects for future development by Fatoumata Ousmane Maiga, Laurent Dembele, Mohamed Maiga and Abdoulaye A. Djimde in Therapeutic Advances in Infectious Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.