
Abstract
Background
Familial hypercholesterolemia is characterised by high low-density lipoprotein-cholesterol levels and is caused by a pathogenic variant in LDLR, APOB or PCSK9. We investigated which proportion of suspected familial hypercholesterolemia patients was genetically confirmed, and whether this has changed over the past 20 years in The Netherlands.
Methods
Targeted next-generation sequencing of 27 genes involved in lipid metabolism was performed in patients with low-density lipoprotein-cholesterol levels greater than 5 mmol/L who were referred to our centre between May 2016 and July 2018. The proportion of patients carrying likely pathogenic or pathogenic variants in LDLR, APOB or PCSK9, or the minor familial hypercholesterolemia genes LDLRAP1, ABCG5, ABCG8, LIPA and APOE were investigated. This was compared with the yield of Sanger sequencing between 1999 and 2016.
Results
A total of 227 out of the 1528 referred patients (14.9%) were heterozygous carriers of a pathogenic variant in LDLR (80.2%), APOB (14.5%) or PCSK9 (5.3%). More than 50% of patients with a Dutch Lipid Clinic Network score of ‘probable’ or ‘definite’ familial hypercholesterolemia were familial hypercholesterolemia mutation-positive; 4.8% of the familial hypercholesterolemia mutation-negative patients carried a variant in one of the minor familial hypercholesterolemia genes. The mutation detection rate has decreased over the past two decades, especially in younger patients in which it dropped from 45% in 1999 to 30% in 2018.
Conclusions
A rare pathogenic variant in LDLR, APOB or PCSK9 was identified in 14.9% of suspected familial hypercholesterolemia patients and this rate has decreased in the past two decades. Stringent use of clinical criteria algorithms is warranted to increase this yield. Variants in the minor familial hypercholesterolemia genes provide a possible explanation for the familial hypercholesterolemia phenotype in a minority of patients.
Keywords
Introduction
Familial hypercholesterolemia (FH) is a common autosomal dominantly inherited disease with a prevalence of 1:250, 1 hallmarked by elevated low-density lipoprotein (LDL) cholesterol levels with a concomitant high risk of premature cardiovascular disease (CVD). 2 FH is diagnosed on the basis of clinical criteria (e.g. Dutch Lipid Clinic Network (DLCN) criteria), ideally followed by DNA sequencing to identify variants in the LDLR, APOB and PCSK9 genes that cause FH. A genetic diagnosis of FH allows better CVD risk stratification, 2 increases therapy adherence of the diagnosed patient 3 and allows cascade testing of first-degree relatives. 4
Apart from the three canonical FH genes, pathogenic variants in other so-called ‘minor FH’ genes may also contribute to or mimic the FH phenotype. 5 Patients with variants in APOE, 6 ABCG5 and/or ABCG8 7 or homozygous variants in LIPA 8 or LDLRAP1 9 often present with hypercholesterolemia. Advances in targeted next-generation sequencing (NGS) methods enabled the sequencing of multiple genes at once, providing genetic information in the classic FH genes, the minor FH genes, as well as other lipid metabolism-related genes in a clinical diagnostic setting. 7 , 10 However, it is yet unknown whether the addition of these extra genes to NGS panels increases the proportion of molecularly diagnosed FH patients.
As diagnostic resources in many countries are limited and FH-causing variants in one of the three culprit genes are not always identified in patients with severe hypercholesterolemia,11–13 the selection of FH patients who are deemed eligible for molecular testing is mostly based on clinical FH criteria such as the DLCN criteria. 14 Moreover, the yield of FH sequencing is proportionally related to the severity of the FH phenotype (i.e. LDL-cholesterol levels). For example, in a general population study with patients with LDL-cholesterol levels above 5 mmol/L only 2% of patients carried a FH-causing variant. 2 This percentage is proportionally associated with the measured LDL-cholesterol level and increases when patients are selected based on additional clinical FH criteria, such as xanthomas, which are incorporated in algorithms such as the DLCN score. Also, Wang and colleagues showed that variants were found in approximately 40% of clinical FH patients with LDL-cholesterol levels between 5 and 6 mmol/L, while approximately 90% of clinical FH patients with LDL-cholesterol levels above 8 mmol/L carried a pathogenic variant in LDLR, APOB or PCSK9. 13
In the present study, we set out to quantify the percentage of patients in whom a pathogenic variant in established and minor FH genes was found, with a targeted NGS approach in a large cohort of patients that was referred to our molecular diagnostics laboratory. Furthermore, we investigated whether, and to what extent, the use of two clinical FH criteria algorithms (DLCN and MedPed) impacted the diagnostic yield and we evaluated the diagnostic yield during the past two decades.
Methods
Referral procedures
The Amsterdam University Medical Center is the national referral centre for DNA diagnostics for patients with suspected genetic dyslipidemias. Physicians who consider a genetic cause to underlie the encountered dyslipidemia in their patient ship a blood sample to the central facility along with a questionnaire containing clinical data about lipid levels, the results of the physical examination, use of medication and the medical and family history of the patient. For this study we analysed data from patients whose referring physician requested molecular FH analysis based on their clinical judgement.
DNA sequencing
Since May 2016, all referrals for genetic dyslipidemia are analysed using an in-house NGS capture covering 27 lipid genes, including LDLR, APOB and PCSK9 (SeqCap easy choice; Roche NimbleGen Inc., Pleasanton, USA). A full list of this gene panel is provided in Supplementary Table 1. Two trained molecular geneticists assessed all variants in the exons and at least 20 base pairs in the adjacent introns of these sequenced genes for their pathogenicity according to standard guidelines for variant classification of the American College of Medical Genetics and Genomics. 15 Detection of copy number variants was based on normalised depth of coverage analysis as its principal method. Using this scoring system, class 4 (likely pathogenic) and class 5 (pathogenic) variants in LDLR, APOB and PCSK9 were considered to be causal. In addition, we investigated class 4 and 5 variants in ‘minor FH genes’, comprising LDLRAP1, APOE, LIPA, ABCG5 and ABCG8. 5 We excluded STAP1 variants from this analysis, as this is no longer considered a FH gene. 16 , 17 Next, the allele frequency of variants in the minor FH genes in FH mutation-negative patients was numerically compared with the allele frequency reported in the European non-Finnish population in gnomAD V2.1.1. (https://gnomad.broadinstitute.org/). 18 The E2/E2 genotype was excluded from this analysis, because this causes dysbetalipoproteinemia which is a distinct phenotype from FH. All variants classified as class 4 and class 5 are hereafter referred to as pathogenic variants.
Before May 2016 (n = 20,912), a molecular diagnosis of FH was made by means of Sanger sequencing of LDLR, APOB and PCSK9 and, when negative with LDL-cholesterol levels above age-specific thresholds, 19 subsequent analysis for copy number variants in LDLR by multiplex ligation-dependent probe amplification.
Patient selection
All consecutive patients who were referred for genetic FH testing, who gave written informed consent and were analysed for variants between May 2016 and July 2018, were eligible for this analysis. Patients were excluded when the LDL-cholesterol level was below 5 mmol/L (cut-off for a DLCN score of at least ‘possible FH’), when data on LDL-cholesterol were missing or reported LDL-cholesterol was obtained during active lipid-lowering treatment. Patients with triglyceride levels greater than 4.5 mmol/L were excluded to prevent enrolling patients with unreliable LDL-cholesterol levels. For the comparison with historical data on Sanger sequencing, we selected all referred patients that were analysed for FH variants between 1999 and May 2016 and compared the data to the NGS yield for all patients with an indication for genetic FH diagnostics, including those with LDL-cholesterol levels below 5 mmol/L. All included patients referred for genetic analysis are index cases. Genetic analysis for cascade screening of members of families with a proved FH variant is performed in a separate program in which targeted variant identification is performed in these individuals. FH patients with homozygous or compound heterozygous pathogenic variants in LDLR, APOB or PCSK9 were excluded from all analyses, except from the overall frequency tables (Table 1, Supplementary Table 2).
FH genotype frequencies.
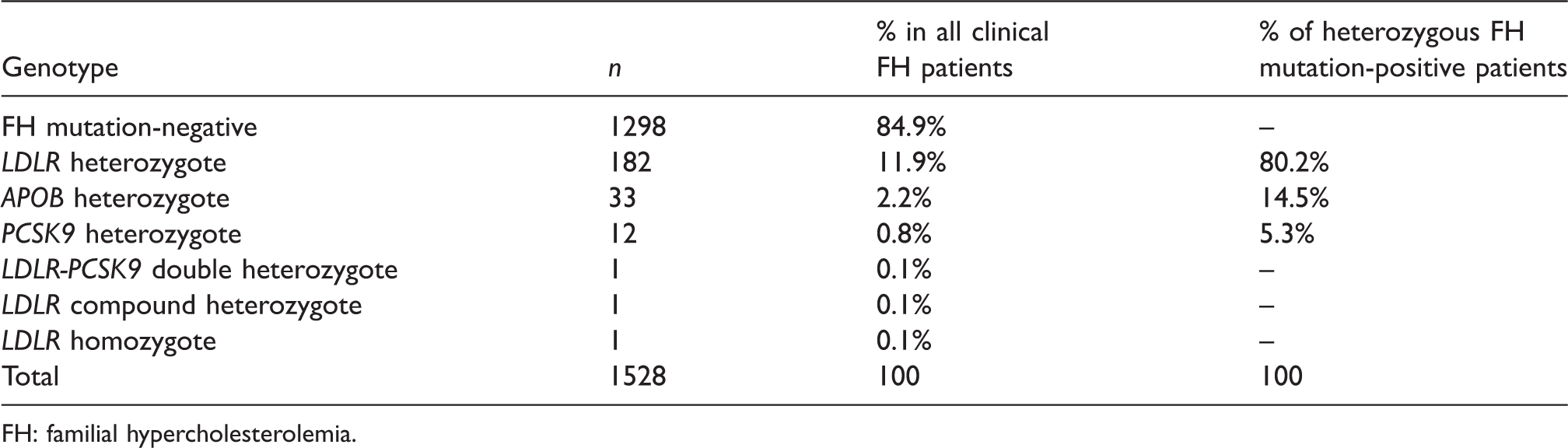
FH: familial hypercholesterolemia.
Clinical FH scoring
We applied two clinical scoring systems for FH. First, as collected data were limited, the Dutch lipid clinical network criteria 14 were modified to fit the collected clinical data (Supplementary Table 5) and applied to the FH cohort. Second, the MedPed criteria for LDL-cholesterol levels in the general population were applied (Supplementary Table 6). 20
Statistical analysis
Normally and non-normally distributed data were compared between groups using independent t-tests and Mann–Whitney U tests, respectively. Categorical data were compared using the Fisher exact test. For analysis of the risk for carrying a FH-causing variant over time, the year of sequencing and the age of the patient were used as covariates in a logistic regression model with FH-causing variant status as the outcome (yes/no). The variable year of sequencing was added to the model with the use of a natural spline and age as a categorical variable consisting of age tertiles. A P value less than 0.05 was considered statistically significant. All statistical analyses were performed using R, version 3.6.1 (R Foundation, Vienna, Austria).
Results
Yield of NGS sequencing
Between May 2016 and July 2018, 2320 index patients with clinical FH were referred to our laboratory for molecular analysis. After excluding patients who were using lipid-lowering therapy, had incomplete lipid data or triglyceride levels above 4.5 mmol/L, a total of 1858 patients remained. A total of 1528 of those patients had LDL-cholesterol levels above 5 mmol/L, corresponding to a DLCN score of ‘possible FH’ (Figure 1). Genetic analysis of the three culprit FH genes (LDLR, APOB and PCSK9) showed that 14.9% of these clinical FH patients were heterozygous carriers of pathogenic variants in these genes (Table 1, Figure 2(a)). Three homozygous FH patients were identified (0.2%, Table 1).
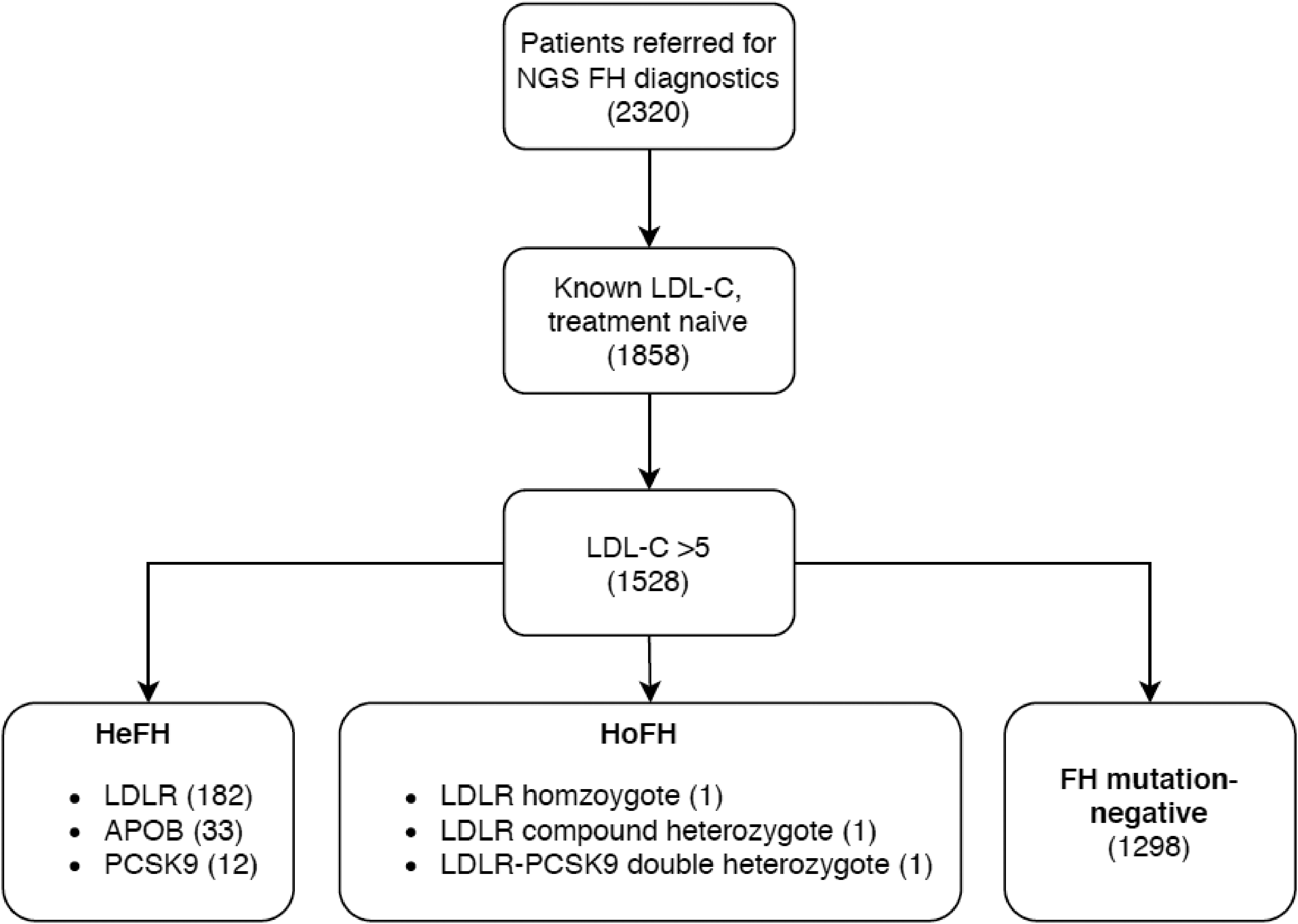
Study design flowchart. Flowchart showing selection of patients included in the analyses. NGS: next-generation sequencing; HeFH: heterozygous familial hypercholesterolemia; HoFH: homozygous familial hypercholesterolemia.
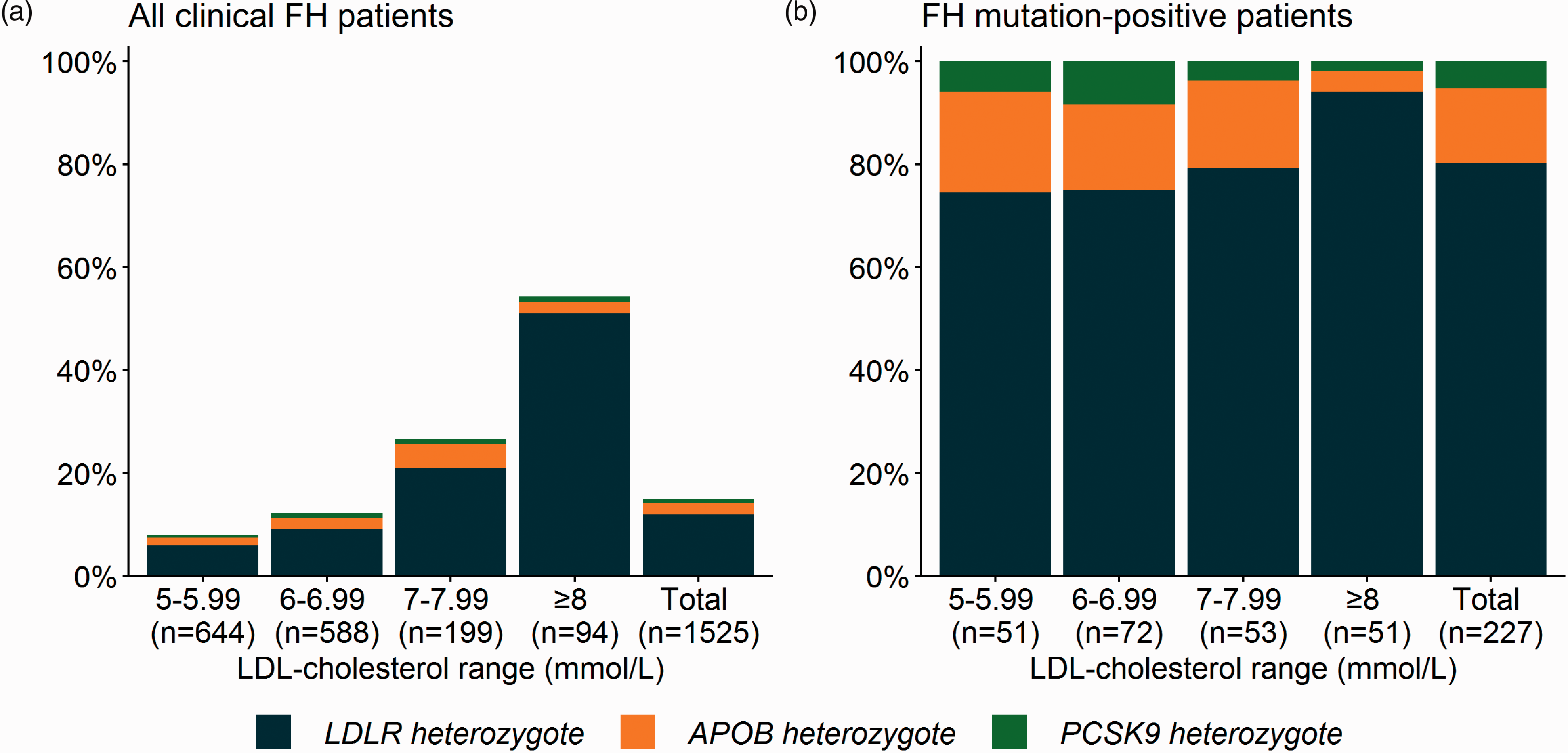
Distribution of FH-causing variants across LDL levels. Proportion of patients with pathogenic variants in LDLR, APOB or PCSK9 stratified according to LDL-cholesterol levels. (a) All clinical FH patients and (b) only patients with genetically confirmed FH. Patients who were found to be homozygote or compound heterozygote for pathogenic variants in LDLR, APOB and PCSK9 were excluded from all graphs. FH: familial hypercholesterolemia; LDL: low-density lipoprotein.
In total, 227 (14.9%) patients were identified who were heterozygous carriers of a pathogenic variant in LDLR, APOB or PCSK9. The proportion of clinical FH patients with such a variant increased with LDL-cholesterol levels and ranged from 7.9% in patients with untreated LDL-cholesterol levels between 5–5.99 mmol/L to 54.3% in patients with LDL-cholesterol levels above 8 mmol/L (Figure 2(a)). In the vast majority of FH mutation-positive patients, LDLR variants were identified (80.2%, Table 1, Figure 2(b)), followed by APOB variants and PCSK9 variants in 14.5% and 5.3% of FH mutation-positive patients, respectively. With increasing LDL-cholesterol levels, the proportion of FH mutation-positive patients with a LDLR variant increased from 74.5% to 94.1% (Figure 2(b)). The distribution of LDL-cholesterol levels for carriers of pathogenic LDLR, PCKS9 and APOB variants is shown in Supplementary Figure 1, and the mutation type frequencies and associated LDL-cholesterol levels are reported in Supplementary Table 4 and Supplementary Figure 2.
Clinical characteristics of FH mutation-positive and FH mutation-negative patients
Genetically confirmed FH patients were younger (mean age 43.6 ± 16.4 vs. 54.5 ± 11.7 years, P<0.001) and had higher total cholesterol (9.2 ± 1.6 vs. 8.4 ± 1.0 mmol/L, P<0.001) and LDL-cholesterol levels (7.1 ± 1.4 vs. 6.2 ± 0.9 mmol/L, P<0.001) compared to those without a pathogenic variant in one of the three FH genes (Table 2). The prevalence of CVD of the patients and their parents was similar between the two groups, except for peripheral artery disease (PAD) and stroke, which were observed more frequently in FH mutation-negative patients (4.5 vs. 1.0%, P = 0.026 and 5.0 vs. 1.4%, P = 0.031, respectively). Furthermore, a history of liver enzyme abnormalities and the use of hormone replacement therapy were significantly more prevalent in FH mutation-negative compared with FH mutation-positive patients (6.6 vs. 2.1%, P = 0.027 and 8.2 vs. 4.7%, P = 0.049, respectively, see Table 2).
Clinical characteristics study population.
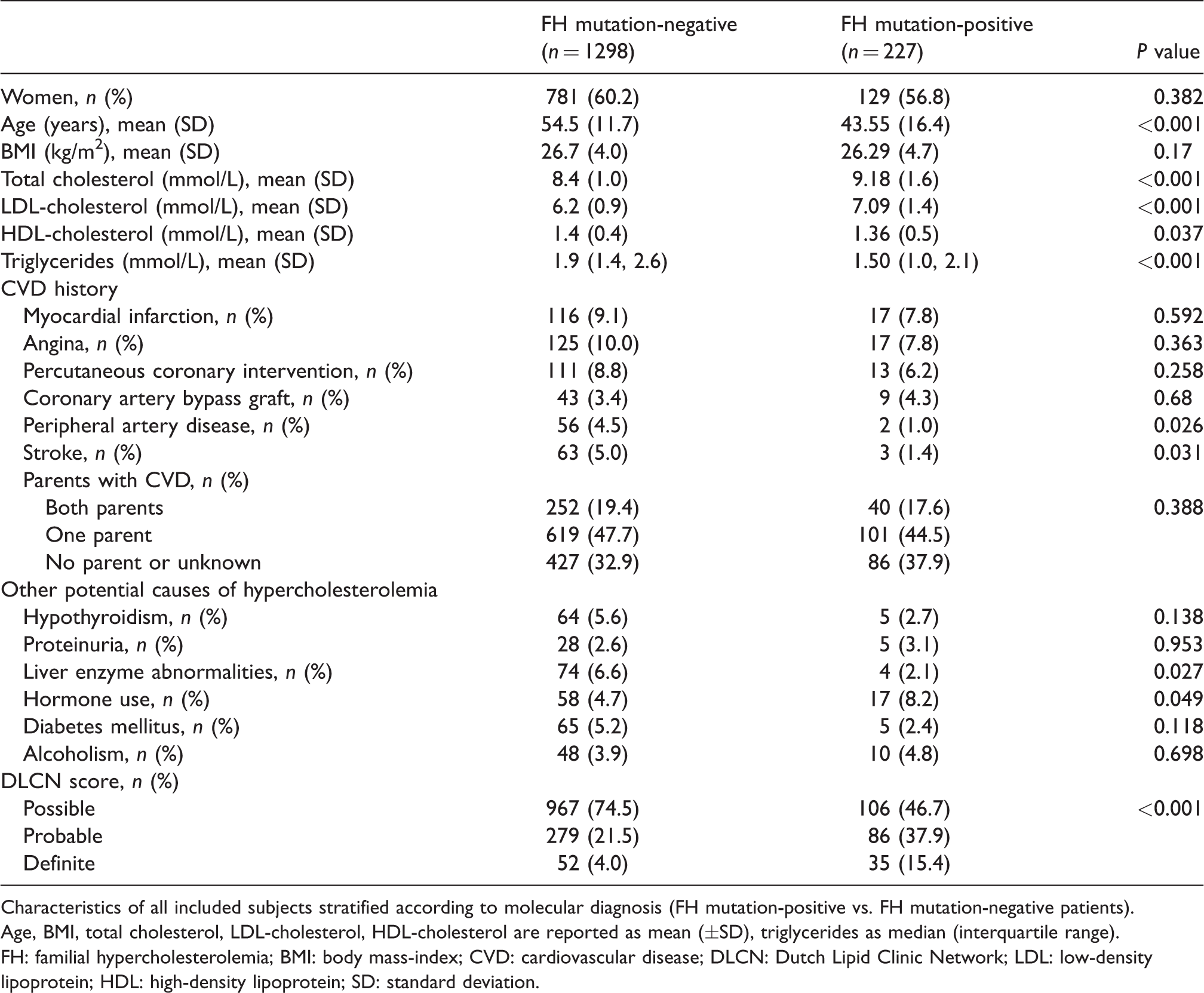
Characteristics of all included subjects stratified according to molecular diagnosis (FH mutation-positive vs. FH mutation-negative patients).
Age, BMI, total cholesterol, LDL-cholesterol, HDL-cholesterol are reported as mean (±SD), triglycerides as median (interquartile range).
FH: familial hypercholesterolemia; BMI: body mass-index; CVD: cardiovascular disease; DLCN: Dutch Lipid Clinic Network; LDL: low-density lipoprotein; HDL: high-density lipoprotein; SD: standard deviation.
Minor FH genes
Apart from the three major genes, variants in a number of other genes have been described to result in a phenotype that mimics FH (i.e. APOE, ABCG5, ABCG8, LIPA, LDLRAP1 are implied to cause FH). We therefore investigated how many patients were carriers of pathogenic variants in these genes. Of the FH mutation-negative patients, 4.8% (n = 63), which equals 4.1% of all patients (excluding those with a combination of a pathogenic variant in both a minor FH gene and in LDLR, APOB or PCSK9) were heterozygous carriers of a pathogenic variant in one of these genes. Eight were true dysbetalipoproteinemia patients with the E2/E2 genotype and two had hypercholesterolemia associated with the recessive disease sitosterolemia (one ABCG8 true homozygote and one ABCG8 compound heterozygote; see Supplementary Table 2). A total of 16 patients had a combination of a FH-causing variant in LDLR, APOB or PCSK9 with one or more homozygous or compound heterozygous variants in the minor FH genes (see Supplementary Table 2). The allele frequency of variants in APOE, ABCG5 and ABCG8 was numerically higher in FH mutation-negative patients compared to a European, non-Finnish, reference population in gnomAD (see Supplementary Table 3). An additional eight variants, not reported in this gnomAD population, were identified. None of the patients were found to be homozygous or compound heterozygous for pathogenic variants in LDLRAP1 or LIPA.
Clinical FH criteria
The DLCN and MedPed criteria are often used in clinical practice to diagnose FH. We investigated whether the proportion of patients with a FH-causing variant increased with the use of clinical FH criteria (see Supplementary Tables 5 and 6). While we found that 14.9% of the subjects in our cohort carried a variant in the culprit genes, this percentage was 9.9%, 23.6% and 40.2% when patients were stratified for possible, probable and definite FH according to the modified DLCN criteria, respectively (Figure 3(a), Supplementary Table 7). These percentages increased to 25.0%, 50.9% and 50.0% when the modified DLCN criteria were only applied to patients with complete data for DLCN criteria (n = 219, Figure 3(b), Supplementary Table 7). A total of 32.8% of the patients classified as having FH using the MedPed criteria had genetically confirmed FH (Figure 3(c), Supplementary Table 8).
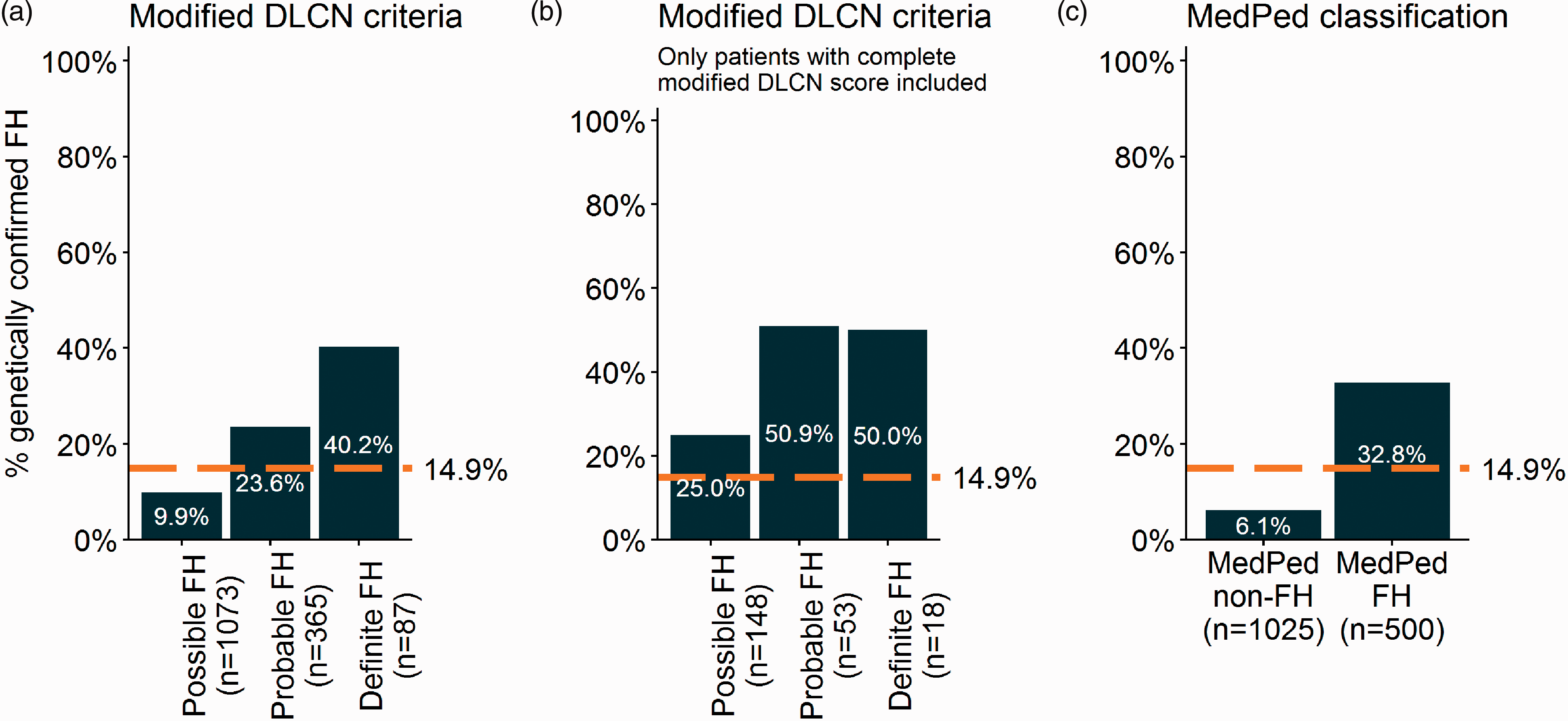
Percentage of patients with genetically confirmed FH per clinical criteria categories. Proportion of patients with genetically confirmed FH stratified according to clinical FH diagnosis: (a) modified DLCN criteria; (b) modified DLCN criteria, only included patients with complete data; (c) MedPed classification. Patients who were found to be homozygote or compound heterozygote for pathogenic variants in LDLR, APOB, PCSK9 were excluded from all graphs. FH: familial hypercholesterolemia; DLCN: Dutch Lipid Clinic Network.
Yield of genetic sequencing over time
Next, we evaluated the yield of genetic FH testing in The Netherlands during the past two decades. Figure 4 and Supplementary Figure 3 show that the percentage of genetically confirmed FH patients per year has decreased since 1999. We observed that this decrease over time differed significantly between age tertiles (see Supplementary Table 9); patients in the lowest tertile (younger than 44 years) were most likely to carry a FH-causing variant, but also showed the largest decrease in yield over time from 40.4% in 1999 to 19.5% in 2018. Overall, the average age of the sequenced clinical FH patients increased from 44.1 ± 14.5 to 51.3 ± 14 years during this period (see Supplementary Figures 4 and 5).
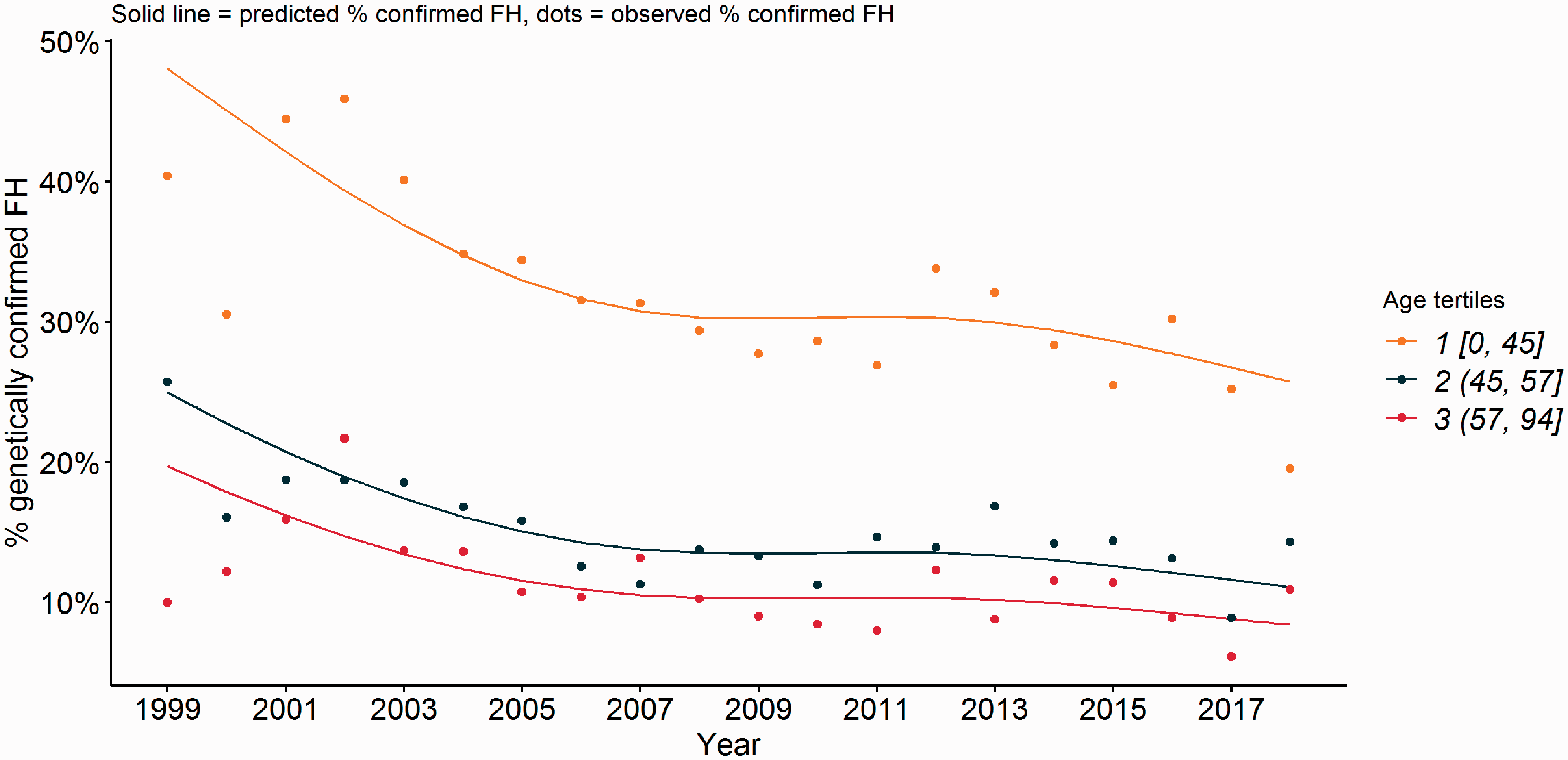
Predicted and observed risk of genetically confirmed FH as a function of year and age. Predicted and observed risk of genetically confirmed FH as a function of year and age. Analysis was performed with a logistic regression analysis with the natural spline of year and age tertiles as covariates. The solid line represents the predicted risk (or proportion) of genetically confirmed FH patients over time. Dots represent the observed proportion of genetically confirmed FH in a given year. FH: familial hypercholesterolemia.
Discussion
We investigated the diagnostic yield of NGS in a large number of patients referred for molecular testing for FH variants. A FH-causing genetic variant was identified in only 14.9% of FH patients with LDL-cholesterol levels of 5 mmol/L or greater. This percentage increased to more than 50% when patients were stratified according to either higher LDL-cholesterol levels or more stringent diagnostic FH criteria ascertained by the DLCN criteria; 4.8% of FH mutation-negative patients were heterozygous carriers of a pathogenic variant in a minor FH gene. When comparing the yield of NGS for FH with historical Sanger sequencing data, we found that the likelihood of finding a pathogenic FH variant has decreased from 30% in the early 2000s to approximately 15% in the past few years. This decrease was largely due to a decline in the prevalence of variants in younger patients.
An average genetic yield of approximately 15% in our cohort is relatively low compared to previous studies. A Canadian study with 313 patients reported that 42% and 88% of patients with LDL-cholesterol between 5 and 6 mmol/L and above 8 mmol/L, respectively, carried a FH-causing variant. 13 A more recent study from the same research group found that 38% of 924 patients with a ‘FH phenotype’ (typically LDL ≥5 mmol/L) who were referred for NGS carried a pathogenic FH-causing variant. 12 In an unrelated Canadian FH cohort, a pathogenic FH variant was identified in 275 of 626 patients (44%) with clinically diagnosed FH, 21 while 30% of the patients were found to be mutation-positive in an Australian cohort of adult patients with at least possible FH. 22 In the Italian LIPIGEN study, 1592 patients with probable or definite FH according to the DLCN criteria were sequenced using NGS and 1076 (68%) were found to carry a pathogenic FH mutation. 23
Our lower yield probably reflects the mode of referral that led to our cohort and might be caused by the fact that NGS diagnostics is accessible to a heterogeneous group of referring physicians in The Netherlands (e.g. general practitioners, cardiologists, internists) as opposed to a request for NGS analysis in specified lipid clinics in other countries. 12 , 13 , 22 In addition, NGS analysis costs are covered by the basic health insurance of all Dutch inhabitants, and this unrestricted access may lower the threshold for requesting molecular testing compared to regions where more restrictions apply. In such a situation it is also easier for a physician to use genetic testing as a method to exclude FH.
In our analyses, we included all patients who were referred for sequencing and had a LDL-cholesterol above or equal to 5 mmol/L, regardless of other phenotypic characteristics. However, when we retrospectively applied clinical FH criteria to our dataset, the yield increased, and was larger than the overall observed yield in our study. Thus, more than 50% of patients with a DLCN score indicating ‘probable’ or ‘definite’ FH, for whom we obtained complete data, were carriers of a FH-causing variant. This, however, has to be interpreted with caution because only 219 patients (14.4%) had complete data for DLCN calculation, potentially introducing a bias as forms may have been filled out more completely for patients in whom the referring physician was more confident that a variant would be identified. However, a positive MedPed score, which only requires age and an untreated LDL-cholesterol level for its calculation, also increased the likelihood of identification; 32.8% of the patients with FH based on the MedPed score carried a FH-causing variant. These findings underscore that a strict application of clinical criteria for FH will increase the proportion of patients in whom a variant is identified.
The inclusion of phenotypically less severe hypercholesterolemic patients now may also explain the decline of the yield we observed over time to the current 14.9%, and might be a result of the successful cascade-screening program for FH conducted in The Netherlands between 1993 and 2014. 24 We hypothesise that subjects with frequently occurring FH-causing variants had already been identified in the early years of the program, leaving the remainder of the population enriched for hypercholesterolemia of other origins, especially in subjects with lower LDL-cholesterol levels compared to those with extremely elevated LDL-cholesterol levels. Unfortunately, we cannot confirm this screening success hypothesis with the current data.
Despite having a clinical FH diagnosis based on clinical criteria, a relatively large proportion of FH patients remains undiagnosed at a molecular level after NGS sequencing. The FH phenotype in these subjects could be explained by (a combination of) other factors, including: pathogenic (intronic) variants in regions that are not covered by NGS, 25 polygenic hypercholesterolemia, 26 high lipoprotein(a) levels, 27 , 28 mixed dyslipidemia (familial combined hyperlipidemia) 29 or incidental and secondary hypercholesterolemia. 30 Our data did not permit us to rule out these factors in our current cohort of FH patients. Unfortunately, our sequencing capture did not include the single nucleotide polymorphisms to calculate a FH polygenic risk score. It is likely that a significant proportion of FH mutation-negative patients suffers from polygenic hypercholesterolemia as described by others, and that the proportion of polygenic hypercholesterolemia patients has relatively increased compared to monogenic FH over the years. 13 , 26 Furthermore, patients with high lipoprotein(a) levels and incidental hypercholesterolemia have recently been reported to be prevalent in this group as these patients present with moderately elevated LDL-cholesterol levels. 28 , 30 This was illustrated by 2219 healthy blood donors who had FH according to the MedPed criteria, and of whom 71% no longer met the MedPed criteria for FH at a second LDL-cholesterol measurement at a later time point. 30 This clearly illustrates the variability in LDL-cholesterol levels and may cause overestimation of the FH prevalence in cohorts selected based on only one LDL-cholesterol measurement, such as ours. Therefore, it might be beneficial only to request molecular testing for FH when elevated LDL-cholesterol has been observed with multiple measurements.
Apart from the three major FH genes (i.e. LDLR, APOB, PCSK9), additional ‘minor FH genes’ (i.e. APOE, ABCG5, ABCG8 and LIPA) are implied as being associated with the FH phenotype. 5 However, more research on the association between variants in these genes and the occurrence of the FH phenotype is needed. For example, it is still unclear whether variants in ABCG5 or ABCG8 are causative for autosomal dominant FH, or are associated with an overlapping FH phenotype. 7 , 31 Equally challenging are variants in APOE. The E2/E2 APOE genotype is best known to cause the distinct dysbetalipoproteinemia phenotype, which was observed in eight individuals in our cohort and might thus have been clinically misdiagnosed. Furthermore, other APOE variants are implied to be causative for FH instead of dysbetalipoproteinemia. 6 Taken together, pathogenic variants in minor FH genes were found in 4.8% of FH mutation-negative patients and 4.1% of all included patients, and the allele frequency of these variants was higher in FH mutation-negative patients compared to a reference population. However, the degree to which these variants explain the FH phenotype in FH mutation-negative patients remains uncertain. This implies that only a minority of patients might benefit from a genetic diagnosis in one of these genes.
Our study has several strengths. Firstly, our NGS platform offers comprehensive coverage of a large number of genes associated with dyslipidemia. Secondly, our centre is the national referral centre for genetic dyslipidemia and we were therefore able to assemble a large cohort of treatment-naive patients that underwent NGS for the clinical suspicion of FH. However, this study also has several limitations. Although data were obtained through a standardised referral form, correct and complete data entry was entirely dependent on the referring physician. As a consequence, FH stigmata and clinical data may have either been over- or underreported, depending on the level of expertise of the referring physician. In addition, we excluded all patients who were taking lipid-lowering therapy on referral and/or had no known LDL-cholesterol levels, greatly reducing the number of patients for analysis.
In conclusion, we have shown that comprehensive NGS of a large cohort (n = 1528) of patients with a clinical suspicion of FH and LDL-cholesterol levels of 5 mmol/L or greater yielded a molecular diagnosis in almost 15% of the cases. Patients with a pathogenic FH variant were younger, had higher plasma LDL-cholesterol levels and lower triglycerides. Diagnostic yield increased when more stringent selection according to the DLCN score, MedPed criteria, or plasma LDL-cholesterol level was applied. Furthermore, the diagnostic yield decreased over time from approximately 30% in the early 2000s to approximately 15% now, and this decline was most salient in younger patients with suspected FH. The relatively low and declining diagnostic yield advocates the more stringent preselection of clinical FH patients before diagnostic NGS is performed.
Supplemental Material
sj-pdf-1-cpr-10.1177_2047487320942996 - Supplemental material for Next-generation sequencing to confirm clinical familial hypercholesterolemia
Supplemental material, sj-pdf-1-cpr-10.1177_2047487320942996 for Next-generation sequencing to confirm clinical familial hypercholesterolemia by Laurens F Reeskamp, Tycho R Tromp, Joep C Defesche, Aldo Grefhorst, Erik SG Stroes, G Kees Hovingh and Linda Zuurbier in European Journal of Preventive Cardiology
Footnotes
Author contribution
LFR and GKH contributed to the conception or design of the work. All authors contributed to the acquisition, analysis, or interpretation of data for the work. LFR and TRT drafted the manuscript. JCD, AG, ESGS, GKH and LZ critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work ensuring integrity and accuracy.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GKH has served as consultant and speaker for biotech and pharmaceutical companies that develop molecules that influence lipoprotein metabolism, including Regeneron, Pfizer, MSD, Sanofi and Amgen. Until April 2019 GKH served as PI for clinical trials conducted with Amgen, Sanofi, Eli Lilly, Novartis, Kowa, Genzyme, Cerenis, Pfizer, Dezima and Astra Zeneca; and with current and past research grants from ZonMW (ViDi 016.156.445), EU, AMGEN, Sanofi, AstraZeneca, Aegerion and Synageva. The Department of Vascular Medicine, Amsterdam University Medical Center, receives honoraria and investigator fees for sponsor driven studies/lectures for companies with approved lipid-lowering therapies in The Netherlands. Since April 2019, GKH is partly employed by Novo Nordisk (0.7FTE) and the AMC (0.3FTE). ESGS reports personal fees from Novartis, personal fees from Amgen, personal fees from Sanofi-Regeneron, personal fees from Mylan, personal fees from Esperion, unrelated to the submitted work. All fees were paid to the institution.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by a ZonMW grant (VIDI no. 016.156.445) obtained by GKH.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.