
Abstract
The upregulation of Sphingosine kinase 1 (SphK1) expression and accompanied sphingosine-1-phosphate (S1P) production have been reported to contribute to the proliferation of pulmonary artery smooth muscle cells (PASMC) and pulmonary arterial remodeling. However, the molecular mechanisms of SphK1/S1P upregulation in PASMC and the specific mechanisms of how SphK1/S1P pathway promotes PASMC proliferation remain largely unclear. This study aims to address these issues. Here, we demonstrated that TGF-β1 significantly upregulated SphK1 expression and S1P production by promoting the phosphorylation of Smad2/3 in PASMC. Further study indicated that SphK1/S1P pathway mediated TGF-β1-induced Notch3 activation in PASMC. In addition, we showed that TGF-β1 significantly induced proliferation of PASMC, while pre-inhibition of Smad2/3 phosphorylation with SB431542 or silencing SphK1 using small interfering RNA in advance, or pre-blocking Notch3 pathway with N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), attenuated TGF-β1-induced PASMC proliferation. Taken together, our study indicates that Smad2/3/SphK1/S1P/Notch3 pathway mediates TGF-β1-induced PASMC proliferation and suggests this pathway as a potential therapeutic target in the prevention and treatment of pulmonary hypertension.
Introduction
Pulmonary hypertension (PH) is a fatal disease characterized with functional and structural changes of pulmonary vasculature, which consequently leads to the increase of pulmonary vascular resistance (PVR) and elevation of pulmonary arterial pressure (PAP), and finally results in right heart failure and death. 1 Different types of PH share similar pathogenesis including sustained vasoconstriction, uncontrolled pulmonary vascular remodeling, and thrombosis in situ. 2 Hyperproliferation of pulmonary artery smooth muscle cells (PASMC) is the most significant feature of pulmonary vascular remodeling. 3 The precise mechanisms of PASMC hyperproliferation are poorly understood and few effective treatments are available. 4 Therefore, understanding the molecular mechanistic regulation of PASMC proliferation in PH is essential for developing novel therapeutics.
Sphingosine kinase 1 (SphK1) is a highly conserved lipid kinase for phosphorylating sphingosine to generate sphingosine-1-phosphate (S1P). 5 Increased expression of SphK1 and production of S1P have been reported to regulate diverse cellular processes including cell proliferation, differentiation, migration, and survival.6,7 It has been found that levels of SphK1 and S1P are elevated in patients with PH and genetic deletion or pharmacologic inhibition of SphK1 protects from the development of experimental PH in several rodent models.8,9 Studies have shown that SphK1 can be stimulated by numerous external stimuli, such as growth factors and their receptors, including platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), tumor necrosis factor α (TNF-α), and transforming growth factor β (TGF-β). 5 TGF-β has been implicated in the pathogenesis of PH, the expression of TGF-β is increased in pulmonary arteries and lungs of PH patients and animal models, and TGF-β blockade can prevent PH in multiple experimental models.10–13 Activation of Smad signaling pathway has been demonstrated to be predominantly responsible for TGF-β-induced proliferation of multiple cell types. 14 Further studies have found that Smad2/3 mediates TGF-β1-induced SphK1 expression in myofibroblasts. 15 However, it is still unknown whether TGF-β1 can upregulate the expression of SphK1 by activating Smad2/3 pathway in PASMC.
Notch is a group of transmembrane receptors composed of four members (Notch 1–4); the ligand of Notch consists of five members (JAGGED 1/2 and DELTA 1/3/4). 16 Upon ligand binding, the Notch receptor is activated by two sequential steps of proteolytical cleavage to release the intracellular domain of the Notch receptor (NICD). 17 NICD translocates into the nucleus and activates the DNA-binding factors to regulate the transcription of specific target genes. 17 Recent studies have shown that Notch3 signaling pathway is involved in the development of PH by promoting the proliferation of PASMC.18,19 Hirata et al. have reported that SphK1/S1P promotes proliferation of breast cancer cells via activating Notch signaling pathways. 20 Further investigation is needed as to whether SphK1/S1P mediates activation of Notch3 signaling pathway in PASMC.
To explore the above issues, we inhibited Smad2/3, SphK1/S1P, and Notch3 signaling pathways in primary cultured PASMC stimulated with TGF-β1, respectively. We found that TGF-β1 upregulated SphK1 expression and S1P production by activating Smad2/3 pathway, SphK1/S1P further mediated the activation of Notch3 signaling pathway and ultimately mediated TGF-β1-induced PASMC proliferation.
Materials and methods
Cell preparation and culture
Primary PASMC were obtained from pulmonary arteries of Sprague-Dawley male rats (70–80 g) according to the method reported previously.21–24 All animal care and experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of Xi’an Jiaotong University Animal Experiment Center. All protocols used in this study were approved by the Laboratory Animal Care Committee of Xi’an Jiaotong University. In brief, pulmonary arteries were rapidly isolated from sacrificed rats, washed in phosphate buffered saline (PBS; 4℃). The adventitia was gently stripped off with forceps and the endothelium was carefully removed by scratching the intima surface with an elbow tweezers. The remaining smooth muscle layer was cut into 1-mm 3 tissue blocks and transferred into a culture flask with Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Isle, NY, USA) containing 10% fetal bovine serum (FBS; Sijiqing, HangZhou, China) and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin), and incubated at 37℃ in a humidified 5% CO2 incubator. Cells were passaged at 80% confluence using 0.25% trypsin (Invitrogen, Carlsbad, CA, USA). Cells between passages 2–6 were used for further experiments. The purity of PASMC was confirmed by immunostaining with α-smooth muscle actin (α-SMA; Sigma, St. Louis, MO, USA) as described previously. 25 Fluorescence microscope images showed that >90% of the cells are smooth muscle cells (data not shown here). Before each experiment, cells were serum-starved (1% FBS in DMEM) overnight to minimize serum-induced effects. TGF-β1 (Peprotech, Rocky Hill, NJ, USA) was used to stimulate PASMC. SB431542 (Cell Signaling Technology, Beverly, MA, USA), a specific inhibitor of the TGF-β type 1 receptors ALK5, was used to inhibit phosphorylation of Smad2/3. 26 DAPT (Santa Cruz Biotechnology, Dallas, TX, USA), a γ-secretase inhibitor, was employed to block Notch pathway.19,27
siRNA transfection
To silence the expression of SphK1 protein, PASMC were transfected with sequence-specific (5′-GACGGCAACUCUAUUCUGUTT-3′) or non-targeting control (5′-UUCUCCGAACGUGUCACGUTT-3′) siRNA (GenePharm, Shanghai, China) using Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols. Briefly, PASMC were seeded into six-well plates and cultured until reaching 30–50% confluence; 100 nM siRNA and 5 µL Lipofectamine were separately diluted in 250 µL serum-free DMEM and incubated for 5 min at room temperature. Next, the diluted siRNA was gently mixed with the diluted Lipofectamine and incubated at room temperature for 20 min. Then, the complex of siRNA and Lipofectamine was added into cells, and the cells were incubated in serum-free DMEM containing the siRNA and Lipofectamine complex for 6 h. Then, the medium was replaced with fresh DMEM containing 10% FBS and cells were cultured for 48 h at 37℃ in a humidified 5% CO2 incubator. Effects of siRNA transfection were analyzed using western blotting.
Cell viability
Cell viability was measured using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. First of all, PASMC were seeded into 96-well plates at 5 × 103 cells/well, allowing to adhere for at least 24 h, and serum starved overnight (1% FBS in DMEM) before the start of experiments. After different treatments, 20 µL MTT solution (5 mg/mL; GenView, FL, USA) was added into each well and the mixture was incubated at 37℃ for an additional 4 h. Finally, 150 µL Dimethyl sulfoxide was added to dissolve the formazan crystals and the reaction product was quantified by measuring the absorbance at 490 nm using a microplate reader (Thermo, Waltham, MA, USA).
ELISA
The levels of S1P were detected strictly in accordance with the instruction of the S1P ELISA kits (Shanghai Westang Bio-tech, Shanghai, China). Briefly, samples were extracted and added to the plate coated with S1P in advance, followed by incubation with HRP labeled antibody for 1 h at 37℃ and five washes of the plate with washing buffer. Then, an equal volume of 3,3′,5,5′-Tetramethylbenzidine was added and incubated again for 15 min at 37℃ in the dark. Next, sulfuric acid was added to stop the reaction. Each sample was repeated three times, the results were measured at 450 nm using a microplate reader (Thermo, Waltham, MA, USA).
Western blotting
The cultured cells were washed twice with ice-cold PBS and then lysed in RIPA lysis buffer containing 50 mM Tris/HCl (pH 7.4), 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), 150 mM NaCl, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride, 1 mM Na3VO4, 1 mM NaF, and proteinase inhibitors. Lysates were centrifuged at 15,000 rpm at 4℃ for 15 min and the liquid supernatants were collected as sample proteins. Proteins were separated on 8–10% SDS-PAGE gel and transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). After blocking for 1 h at room temperature with 5% non-fat milk, the membranes were incubated overnight at 4℃ with primary antibodies against SphK1 (1:1000 dilution; Cell Signaling Technology, USA), p-Smad2/3 (1:800 dilution; Cell Signaling Technology, USA), t-Smad2/3 (1:1000 dilution; Abcam, Cambridge, UK), NICD3 (1:500 dilution; Abcam, Cambridge, UK), and β-actin (1:1000 dilution; Santa Cruz, USA). Membranes were then washed three times in PBST (PBS containing 0.1% Tween-20) for 10 min, followed by incubation at room temperature for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:5000 dilution; Sigma-Aldrich, St. Louis, MO, USA) or goat anti-mouse secondary antibody (1:5000 dilution; Santa Cruz, USA). Immunoreactive bands were visualized using the enhanced chemiluminescence (ECL) kit (Milipore, MA, USA). Images were digitally captured using a ChemiDoc XRS System (Bio-Rad, USA). The band densities were quantified using Quantity One software (Bio-Rad, USA).
Statistical analysis
Statistical analysis was carried out using the SPSS 22.0 software. All data were presented as mean ± standard deviation (SD) and analyzed using nonparametric statistical analysis. P < 0.05 was considered as representing a statistical significance between groups.
Results
TGF-β1 upregulates SphK1 expression in PASMC
To examine whether TGF-β1 upregulates SphK1 expression in PASMC, cells were treated with different concentrations of TGF-β1 at different time points and the protein level of SphK1 was detected using western blotting. As shown in Fig. 1a, TGF-β1 upregulated the expression of SphK1 in PASMC at 24 h, 10 ng/mL TGF-β1 triggered a 1.93-fold increase compared with control. Figure 1b shows the time course of 10 ng/mL TGF-β1 regulation of SphK1 protein level, which increased to 1.92-fold compared with control at the time of 24 h and 1.99-fold at 48 h. These results suggest that TGF-β1 upregulates SphK1 expression in PASMC.
(a) PASMC were treated with different concentration of TGF-β1 in the range of 0–30 ng/mL for 24 h; the protein level of SphK1 was detected using western blotting, β-actin served as a loading control (n = 5 each group). (b) PASMC were exposed to 10 ng/mL TGF-β1 for the indicated times; the protein level of SphK1 was detected using western blotting, β-actin served as a loading control (n = 5 each group). *P < 0.05 vs. control.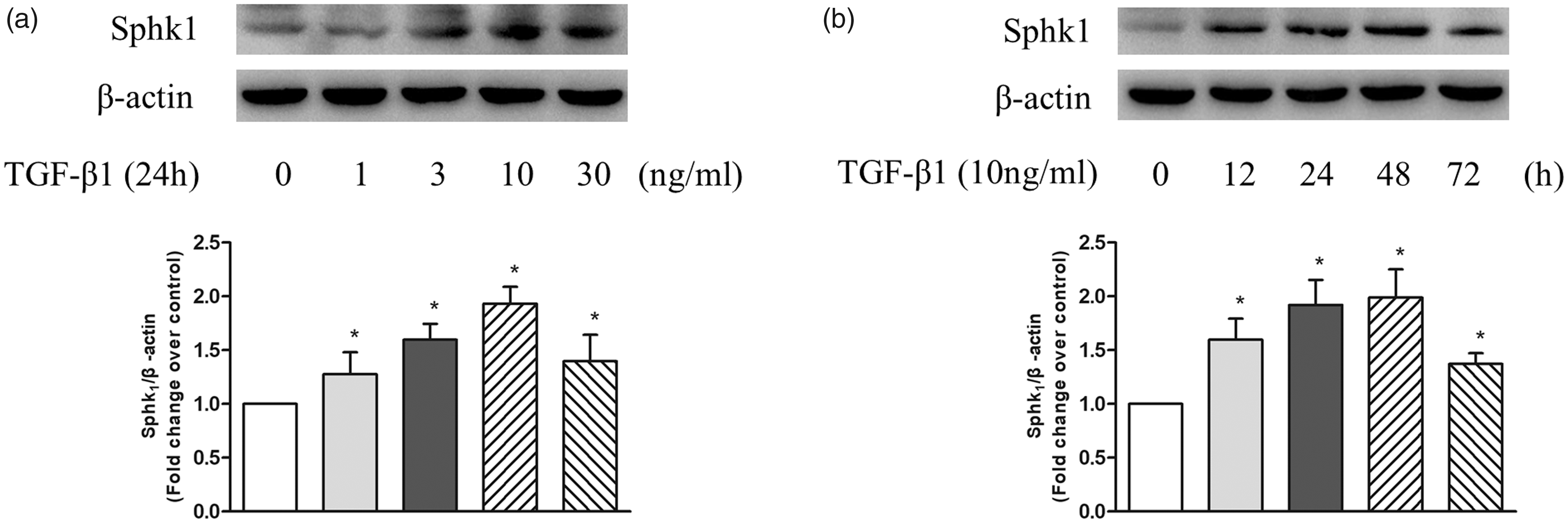
Phosphorylation of Smad2/3 mediates TGF-β1-induced SphK1 expression and S1P production in PASMC
We next investigated whether phosphorylation of Smad2/3 mediated TGF-β1-induced expression of SphK1 in PASMC. PASMC were first incubated with or without 10 μM SB431542 for 1 h to inhibit the phosphorylation of Smad2/3 and followed by stimulation with 10 ng/mL TGF-β1 for 1 h or 24 h, the phosphorylation level of Smad2/3 and the protein level of SphK1 were determined using western blotting.
26
As shown in Fig. 2a, 10 ng/mL TGF-β1 stimulation for 1 h notably increased the phosphorylation level of Smad2/3 in PASMC, while pre-treatment of cells with SB431542 suppressed TGF-β1-induced phosphorylation of Smad2/3. Phosphorylation levels of Smad2/3 reduced from 1.98-fold increase over control in TGF-β1-treated cells to 1.30-fold over control in SB431542 and TGF-β1-treated cells. Figure 2b indicates that 10 ng/mL TGF-β1 stimulation for 24 h resulted in a 2.01-fold increase in SphK1 protein level compared with control, while pre-treatment of cells with SB431542 to inhibit phosphorylation of Smad2/3 reduced SphK1 protein level to 1.49-fold over control in response to TGF-β1. These results suggest that phosphorylation of Smad2/3 mediates TGF-β1-induced SphK1 expression in PASMC.
(a) PASMC were first incubated with or without 10 μM SB431542 for 1 h before stimulation with 10 ng/mL TGF-β1 for 1 h; the protein levels of p-Smad2/3 and t-Smad2/3 were detected using western blotting (n = 5 each group). (b) PASMC were first incubated with or without 10 μM SB431542 for 1 h before stimulation with 10 ng/mL TGF-β1 for 24 h; the protein level of SphK1 was detected using western blotting; β-actin served as a loading control (n = 5 each group). (c) PASMC were first incubated with or without 10 μM SB431542 for 1 h before stimulation with 10 ng/mL TGF-β1 for 24 h; levels of S1P in cell lysates and cell culture supernates were determined using S1P ELISA kits (n = 5 each group). *P < 0.05 vs. control, #P < 0.05 vs. TGF-β1-treated cells.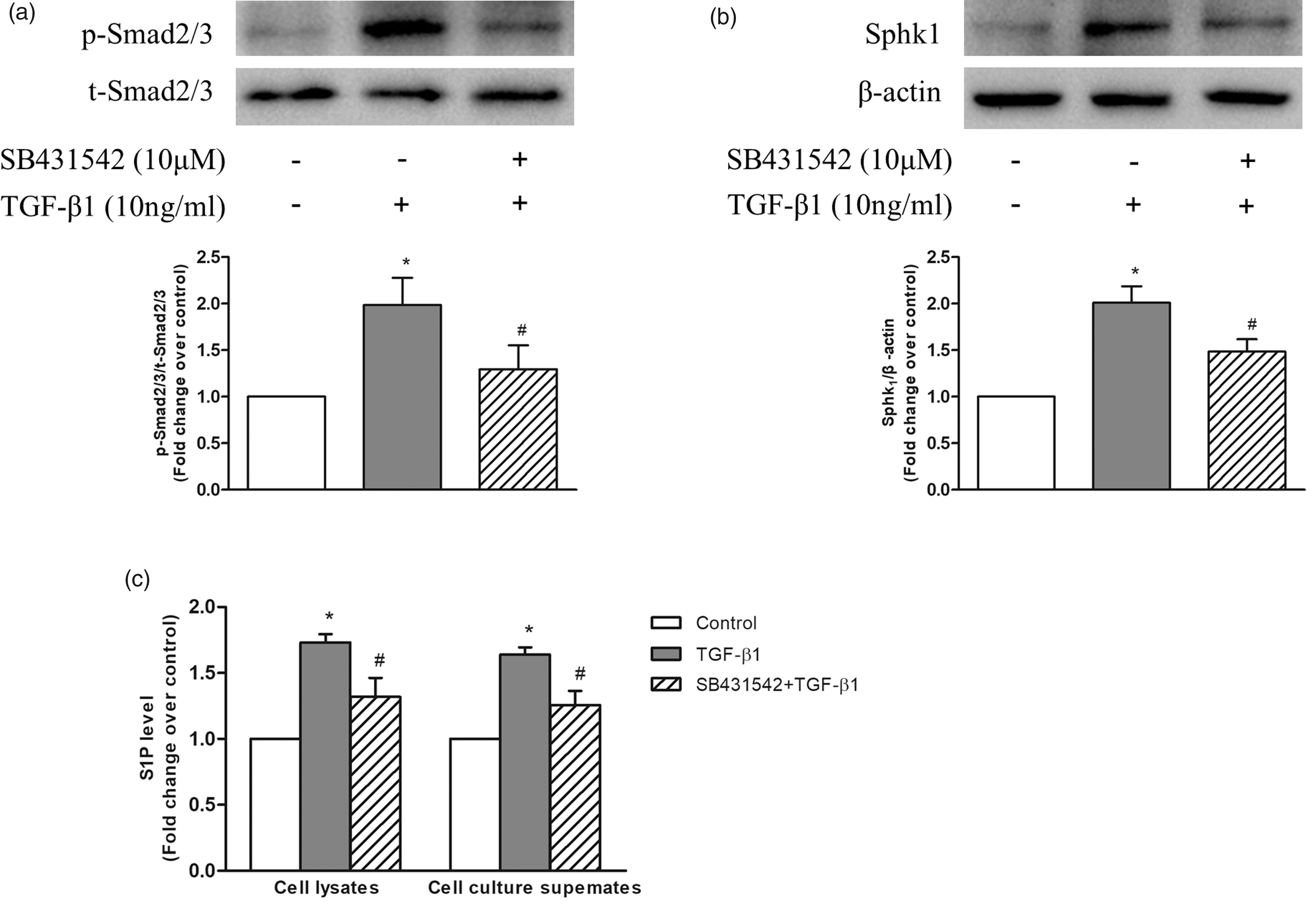
It has been shown that upregulation of SphK1 specifically increases the level of S1P in several types of non-PASMCs. 5 In addition, it has been reported that S1P promotes the proliferation of PASMC.8,9,28 Thus, to clarify whether TGF-β1 upregulates S1P production and phosphorylation of Smad2/3 mediates this process, levels of S1P in cell lysates and cell culture supernates were determined using S1P ELISA kits. PASMC were first treated with or without 10 μM SB431542 for 1 h and then stimulated with 10 ng/mL TGF-β1 for 24 h. As indicated in Fig. 2c, TGF-β1 significantly increased S1P levels to 1.73-fold over control in cell lysates and 1.64-fold over control in cell culture supernates, while pre-treatment of cells with SB431542 to inhibit phosphorylation of Smad2/3 suppressed TGF-β1-induced S1P production to 1.32-fold over control in cell lysates and 1.26-fold over control in cell culture supernates. Together, these results suggest that phosphorylation of Smad2/3 mediates TGF-β1-induced SphK1 expression and S1P production in PASMC.
TGF-β1 promotes Notch3 activation by upregulating SphK1 expression
Two studies have shown that the activation of Notch pathway in non-PASMCs can be induced by S1P or TGF-β,20,29 respectively. To further investigate whether upregulation of SphK1/S1P mediates the effect of TGF-β1 on the activation of Notch3 signaling pathway, siRNA-mediated SphK1 knockdown was performed and the protein level of NICD3 were detected using western blotting. Figure 3a shows that transfection of SphK1 siRNA for 48 h in PASMC reduced SphK1 protein level to 36%, while transfection of non-targeting control siRNA did not affect SphK1 protein level. Figure 3b indicates that 10 ng/mL TGF-β1 stimulation for 24 h significantly increased the protein levels of NICD3, while prior knockdown of SphK1 notably reduced TGF-β1-induced NICD3 levels in PASMC. Protein level of NICD3 reduced from 1.55-fold over control in TGF-β1-treated cells to 1.23-fold over control in SphK1 knockdown cells. These results suggest that TGF-β1 promotes Notch3 activation via upregulating SphK1 expression.
(a) PASMC were transfected with SphK1 sequence-specific siRNA and non-targeting siRNA; SphK1 protein level was determined using western blotting; β-actin served as a loading control (n = 5 each group). (b) PASMC were first transfected with SphK1-specific or non-targeting siRNA for 24 h and then treated with 10 ng/mL TGF-β1 for 24 h; protein level of NICD3 was detected using western blotting, β-actin served as a loading control (n = 5 each group). *P < 0.05 vs. control, #P < 0.05 vs. TGF-β1-treated cells.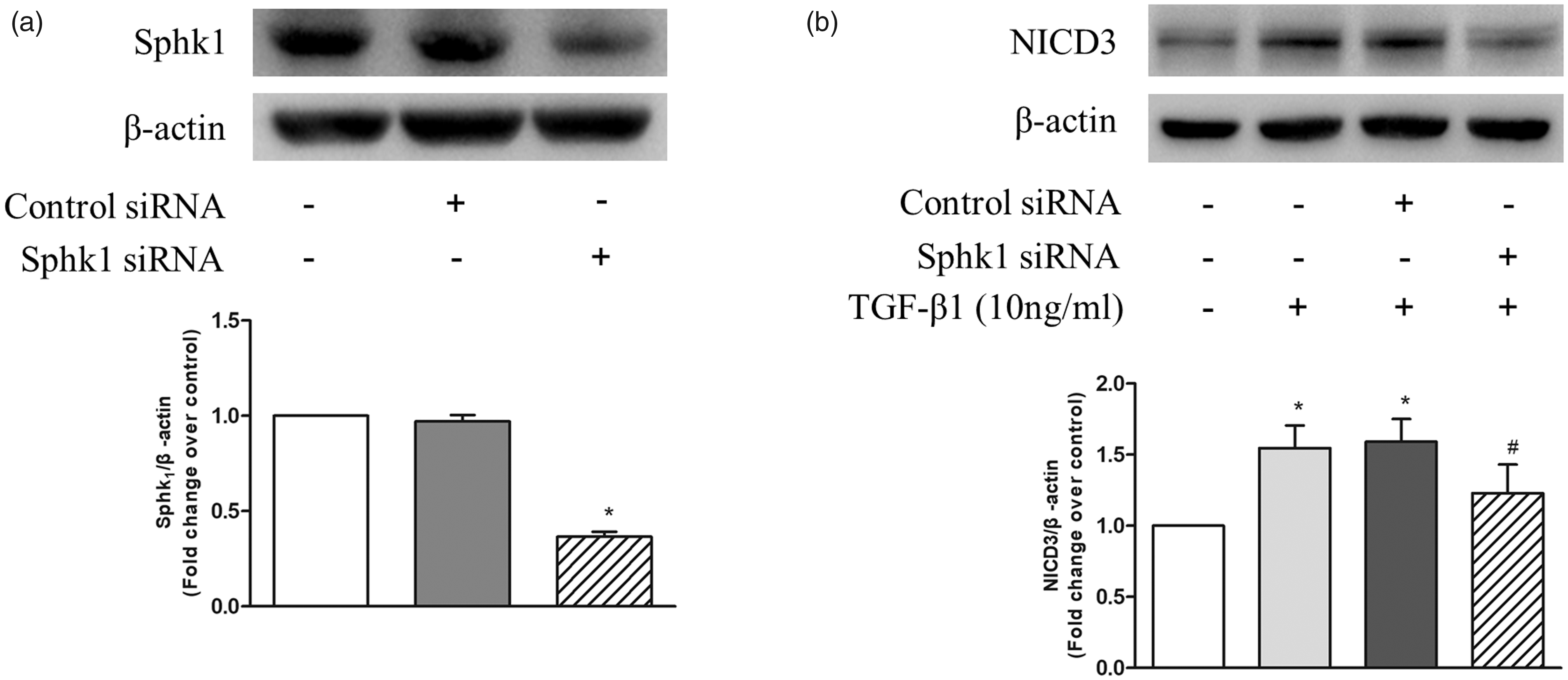
Phosphorylation of Smad2/3, upregulation of SphK1 and subsequent Notch3 activation mediate TGF-β1-induced PASMC proliferation
To clarify whether phosphorylation of Smad2/3, upregulation of SphK1, and subsequent Notch3 activation are involved in TGF-β1-induced PASMC proliferation, cells were pre-incubated with 10 μM SB431542 for 1 h or first transfected with SphK1 siRNA for 24 h or pre-incubated with 10 μM DAPT for 4 h and then stimulated with 10 ng/mL TGF-β1 for an additional 24 h, the proliferation of cells was evaluated by cell viability assay. As shown in Fig. 4, pre-inhibition of Smad2/3 phosphorylation or silencing SphK1 in advance, or pre-blocking the Notch3 pathway, attenuated TGF-β1-induced PASMC proliferation, which was reduced from 1.67-fold over control in TGF-β1-treated cells to 1.27-fold, 0.97-fold, and 1.39-fold over control, respectively. These results suggest that phosphorylation of Smad2/3, upregulation of SphK1, and subsequent Notch3 activation mediates TGF-β1-induced PASMC proliferation.
PASMC were pre-incubated with 10 μM SB431542 for 1 h or prior transfected with SphK1 siRNA for 24 h or pre-incubated with 10 μM DAPT for 4 h and then stimulated with 10 ng/mL TGF-β1 for an additional 24 h; the proliferation of cells was evaluated by MTT (n = 6 each group). *P < 0.05 vs. control, #P < 0.05 vs. TGF-β1-treated cells, &P < 0.05 vs. control siRNA + TGF-β1-treated group.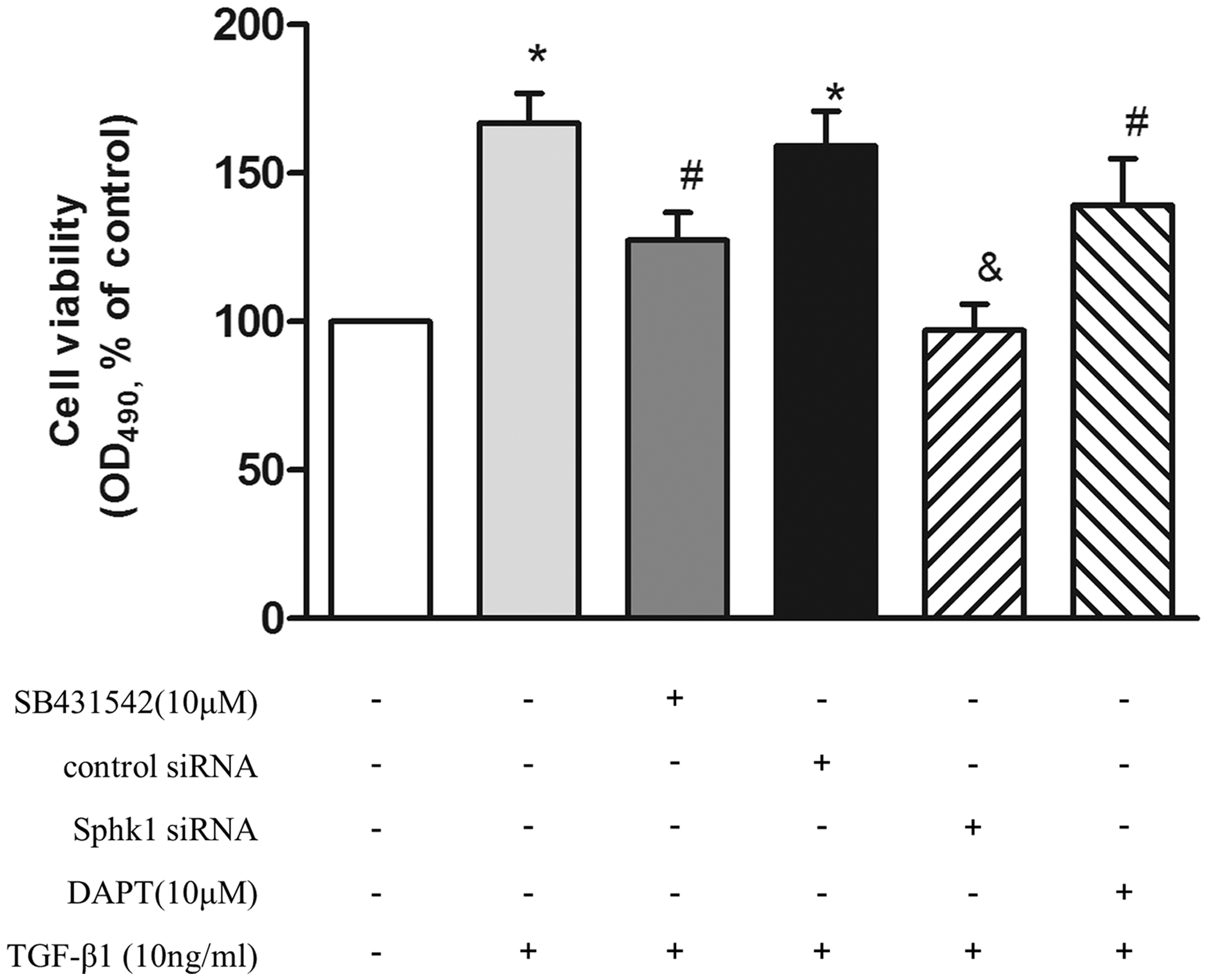
Discussion
PH is a common, fatal disease characterized by increased PVR and elevation of PAP, generally leading to right heart failure and ultimately to death.1,30,31 The precise mechanisms of PH are largely unknown and few effective treatments are available. Due to the key role of PASMC proliferation in the pathogenesis of PH, understanding the mechanistic regulation of PASMC proliferation in PH is crucial for developing new therapeutics. Given the importance of TGF-β1, SphK1, and Notch3 in the pathogenesis of PH and cell proliferation, we investigated potential mechanisms contributing to PASMC proliferation. As outlined in Fig. 5, we demonstrated that TGF-β1 upregulated SphK1 expression and S1P production by promoting phosphorylation of Smad2/3 in primary cultured PASMC, then SphK1/S1P further mediated TGF-β1-induced Notch3 activation, and ultimately mediated TGF-β1-induced PASMC proliferation.
TGF-β1 upregulates SphK1 expression and S1P production by promoting phosphorylation of Smad2/3 in primary cultured PASMC; SphK1/S1P then further mediates TGF-β1-induced Notch3 activation and ultimately mediates TGF-β1-induced PASMC proliferation.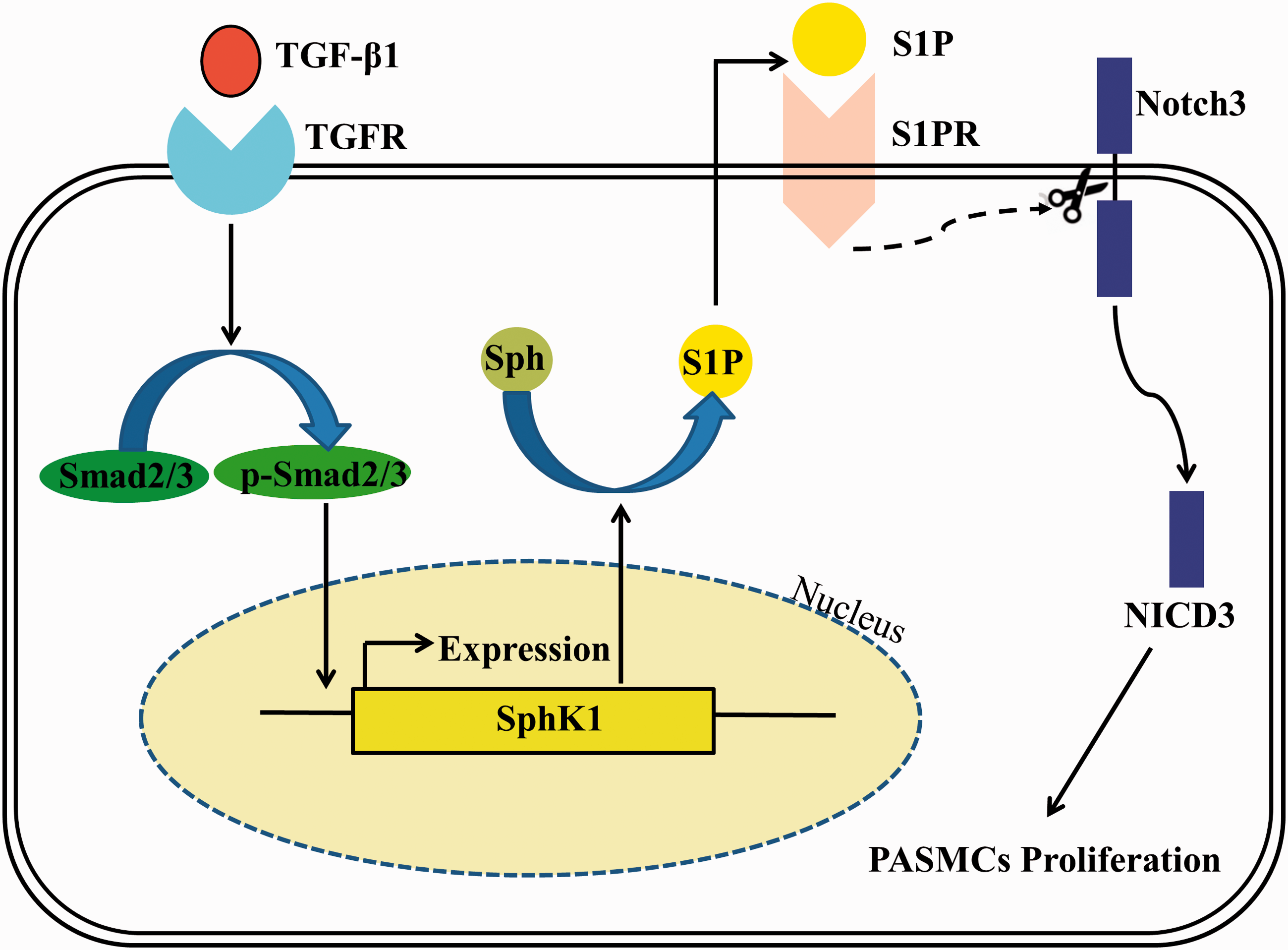
SphK1 is a highly conserved lipid kinase that specifically phosphorylates sphingosine to form S1P. It has been found that SphK1 is widely expressed in liver, kidney, lung, heart, brain, and skeletal muscle. 5 Increased expression of SphK1 and production of S1P have been reported to regulate diverse cellular processes including cell proliferation, differentiation, migration, and survival.6,7 Meanwhile, SphK1 has been reported to be associated with the development of multiple cancer and other human diseases,32–35 while inhibition of SphK1 suppresses the proliferation of both cancer cells and normal cells.8,36,37 Recent studies have shown that SphK1 plays important roles in the pathogenesis and development of PH. Chen et al. and Gairhe et al. have reported that levels of SphK1 and S1P are increased in patients with PH and genetic deletion or pharmacologic inhibition of SphK1 protects from the development of PH in several experimental models.8,9 Sysol et al. have further found that SphK1 mediates PDGF-induced PASMC proliferation. 38 Here, we demonstrated that TGF-β1, an important pathogenic factor of PH, upregulated SphK1 expression and S1P production by promoting phosphorylation of Smad2/3 in primary cultured PASMC, and SphK1/S1P further mediated TGF-β1-induced PASMC proliferation. Similar results have also been found in other cell types, Cencetti et al. have reported that TGF-β1 upregulates SphK1 expression and induces transdifferentiation of myoblasts into myofibroblasts via promoting phosphorylation of Smad2/3. 15 Our study indicates the importance of the TGF-β1/Smad2/3 pathway in activating the SphK1/S1P pathway in primary cultured PASMC and mechanistically linking these important pathways to PASMC proliferation.
Notch3 receptors are mainly expressed in arterial smooth muscle cells and regulate various behaviors of vascular smooth muscle cells including proliferation, apoptosis, and migration.39,40 In the cardiovascular system, it has been demonstrated that Notch3 signaling pathway plays an important role in vascular smooth muscle cells survival and the development of vascular structure, while Notch3-null mice are defective in vascular development. 41 Recent studies have focused on the association between Notch3 and PH. It has been found that Notch3 expression is elevated in patients with PH, and Notch3-knockout mice are resistant to hypoxia-induced PH. 42 Zhang et al. have reported that inhibition of Notch3 reduces proliferation of pulmonary vascular cells and prevents monocrotaline-induced PH by suppressing the expression of Hes1, Skp2, and p27Kip1. 19 On the other hand, Song et al. have proved that overexpression of NICD3 promotes proliferation of PASMC via the Hes1/p27Kip1 signaling pathway. 18 Interestingly, studies have shown that the activation of Notch pathway can be induced by S1P or TGF-β1 in a variety of cells.20,29 However, the detailed association among TGF-β1, S1P, and Notch3 in PASMC is unclear. Here, in our study, we first demonstrated that TGF-β1 promoted Notch3 activation by activating the SphK1/S1P pathway in PASMC.
In conclusion, in this present study, we describe a mechanism responsible for PASMC proliferation in PH and identify the TGF-β1/Smad2/3/SphK1/S1P/Notch3 pathway as a potential therapeutic target in the prevention and treatment of PH.
Footnotes
Acknowledgments
The authors thank the National Local Joint Engineering Research Center for Precision Surgery & Regenerative Medicine, Shaanxi Province Center for Regenerative Medicine and Surgery Engineering Research for its support for this study.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This work was supported by the Natural Science Foundation of China (nos. 81330002 and 81670051).