
Abstract
Artificial intelligence (AI) is transforming medication research and development, giving clinicians new treatment options. Over the past 30 years, machine learning, deep learning, and neural networks have revolutionized drug design, target identification, and clinical trial predictions. AI has boosted pharmaceutical R&D (research and development) by identifying new therapeutic targets, improving chemical designs, and predicting complicated protein structures. Furthermore, generative AI is accelerating the development and re-engineering of medicinal molecules to cater to both common and rare diseases. Although, to date, no AI-generated medicinal drug has been FDA-approved, HLX-0201 for fragile X syndrome and new molecules for idiopathic pulmonary fibrosis have entered clinical trials. However, AI models are generally considered “black boxes,” making their conclusions challenging to understand and limiting the potential due to a lack of model transparency and algorithmic bias. Despite these obstacles, AI-driven drug discovery has substantially reduced development times and costs, expediting the process and financial risks of bringing new medicines to market. In the future, AI is expected to continue to impact pharmaceutical innovation positively, making life-saving drug discoveries faster, more efficient, and more widespread.
Plain language summary
The pharmaceutical industry has enormous and growing amounts of data, and in terms of models, the best AI pharma model is not to build pure AI processes. Combining humans and AI is often superior to human processes or AI processes alone. Just as in chess, the combination of a human and a computer algorithm can usually beat a human or a computer algorithm alone. AI technology methods need to be sorted out and developed. AI’s attention, exploration, and application trials in all sectors of society will inevitably accelerate the maturation and innovation of AI technology methods. When the logic of the “large data → more accurate models → better drugs → more and better data” cycle matures in practice, AI pharma will be significantly accelerated. However, the application and diffusion of any technology are challenging to achieve overnight, and it is the law of development that new things spiral and move in waves. AI and data-driven pharma models need to be explored and practiced more and more before they can truly demonstrate their value.
Introduction
Artificial intelligence (AI) is receiving increasing attention from major pharmaceutical and biotechnology companies worldwide as an engine for new drug development. With three main elements: vast datasets, complex mathematical models, and advanced computational algorithms, AI is a breakthrough in drug discovery and development, bringing new power to the R&D (research and development) of new drugs. Approximately 80% of pharmaceutical and life sciences researchers use AI to accelerate or support their drug discovery efforts. 1 Traditional R&D for new medicines faces many pain points, such as a long cycle, high cost, high failure rate, and low return on investment. AI can potentially improve the efficiency of the new drug development process and the accuracy of predicting drug efficacy and safety, thus increasing the success rate of the drug development pipeline, reducing costs, and shortening the development cycle. AI can provide significant time and cost savings over traditional methods in compound screening and synthesis and substantial savings in clinical trial fees and costs during the clinical research phase. However, there is still some shortsightedness in the application of AI in drug development, such as uneven distribution of data, federated learning not yet widespread, the protection mechanism of algorithm property rights not being refined, and so on. The continuous attention, exploration, and application attempts of pharmaceutical enterprises for AI are bound to accelerate the maturity and innovation of AI pharmaceuticals, which will eventually significantly accelerate.
The history of the development of AI drugs
At the Dartmouth Conference in 1956, computer scientists proposed a new type of computer for intelligence, giving birth to the concept of AI, which refers to the intelligence manifested by machines made by humans and is a new technical science that studies and develops theories, methods, technologies, and applied systems to simulate, extend, and augment human intelligence.1–3 AI has been developed for over 60 years and has successfully moved from theoretical technology to industrial application, leading the way in industry, agriculture, healthcare, finance, etc. 4 AI technology has been developed in autonomous driving, voice recognition, web search, and medical diagnosis. 3 Its ability to perform specific tasks, such as language translation and face recognition, is comparable to or better than that of humans. As a result, it has been commented that “no field is immune to the charms and sweep of AI.” In March 2016, the Al program Alpha Go’s win over the famous South Korean chess player Lee Sedol was a landmark event in the history of AI development. It caused widespread discussion in society. 3
AI has been used in the pharmaceutical field for about 30 years. Since the late 1990s, the underlying algorithmic logic of AI has continued to develop and evolve, experiencing ups and downs (Figure 1). 3 From neural networks to deep neural networks, from machine learning to deep machine learning, continuous optimization iterations of algorithms and accumulation of data have contributed to the development of the entire AI field. 5 From 2001 to the present, the joint development of algorithms, computing power, and data during this period has driven the continuous development of AI pharmaceuticals. Based on different strategies, AI algorithms have enabled different areas of drug discovery, including target discovery, new uses of old drugs, compound screening, molecular design and optimization, protein-protein interactions, crystal shape prediction, dosage form design, ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction, preclinical trial outcome prediction, clinical trial design assistance, patient recruitment, grouping, and many other areas of drug discovery. 3
AI for the pharmaceutical industry.
Since 2018, the development of AI pharma has made the leap from “0” to “1”—the breakthrough from technical concepts to practical application (Figure 2). In the pharmaceutical sector, no AI-enabled drugs have yet been approved for marketing by the FDA (Food and Drug Administration). Still, some AI-enabled pharmaceutical companies have been able to accelerate their Phase I and II clinical drug candidates through AI-enabled approaches. 3 Dozens of AI-enabled drug development pipelines are entering the clinical phase globally. In 2017, reported on research using deep learning (DL) to design drugs for reverse synthetic routes, a breakthrough hailed as the birth of AlphaGo in chemistry. 5 Since then, many industrial companies have made significant breakthroughs in AI-enabled pharmaceuticals. In 2021, Heal-X used AI to find new uses for old drugs, that is, HLX-0201 for fragile X syndrome, advancing the project to Phase II clinical trials within 18 months. 5 In 2019, Deep Genomics used its AI-powered platform to complete novel target discovery and oligonucleotide candidate screening for Wilson’s disease in 18 months. 7 Insilico Medicine applied GENTRL, a generative adversarial network neurotechnology-based approach, to complete an AI drug discovery challenge in 21 days, starting with data collection to build a model to design new molecules, generating the design of a highly active DDR1 (Discoidin Domain Receptor 1) kinase inhibitor. 8 Although the identified compounds exhibit satisfactory microsomal stability and pharmacokinetic properties, further refinement may be necessary to enhance their selectivity, specificity, and other medicinal chemistry characteristics.

An overview of artificial intelligence, machine learning, and deep learning.
DeepMind’s AlphaFold 3 completes a major 50-year biological challenge to predict the 3D structure of proteins. 8 In March 2024, InSys Intelligence’s fully AI-generated drug for idiopathic pulmonary fibrosis (IPF) enters Phase IIa. This is a first-in-class drug with a new backbone compound generated by Chemistry42 (https://pharma.ai/chemistry42). Currently, the AI software PandaOmics is discovering new targets for IPF indications. The successful entry of this drug candidate into the clinic demonstrates the power of AI-enabled innovative drug development. 3 At the same time, it is essential to recognize that analysis conducted using several AI-based methods can be misleading due to overlap between the testing and training sets, biases in the data sets, or results not being analyzed from a chemical perspective. These biases lead to apparently high accuracy, but the general usability of these analyses in prospective research is poor. Undoubtedly, the application of AI in predicting and screening new therapeutic targets and drugs represents an up-and-coming emerging research area. 5 Over the years, there has been a consistent assertion that AI is poised to become a critical tool in expediting drug discovery, development, and testing, thereby serving as a fundamental means to reduce research and testing timelines.
A large number of drugs are under development based on the AI-driven discovery: acute agitation 5 ; acute myelogenous leukemia 5 ; acute-graft-versus-host disease 5 ; adjuvant melanoma 3 ; advanced or metastatic solid tumors 8 ; alopecia 10 ; Alzheimer’s disease 11 ; amyotrophic lateral sclerosis 3 ; atopic dermatitis 3 ; Bronchiolitis obliterans syndrome (BOS), pulmonary sarcoidosis 3 ; breast cancer 5 ; cerebral cavernous 12 ; CMT1A 8 ; COVID-19 8 ; Crohn’s disease 7 ; familial adenomatous polyposis 3 ; familial amyloid polyneuropathy 3 ; FGFR2 5 ; fragile X syndrome 13 ; glioblastoma 3 ; Huntington chorea 13 ; IPF3,12; lung adenocarcinoma 3 ; major depressive disorder, anhedonia 3 ; malformation; Metastatic Castration-Resistant Prostate Cancer (mCRPC) 8 ; metastatic castration-resistant prostate cancer 3 ; metastatic colorectal cancer 7 ; metastatic melanoma 3 ; neurofibromatosis type 2 8 ; oncology 3 ; orexin-1 5 ; phenylketonuria 3 ; PI3Kα 14 ; platinum-resistant ovarian cancer 3 ; psoriatic arthritis 7 ; sarcopenia 15 ; SHP2 10 ; solid tumor 3 ; ulcerative colitis. 12
AI, machine learning, and deep learning
In the CASP14 competition 2020, DeepMind’s AlphaFold2 announced a groundbreaking breakthrough in protein structure prediction with a score well ahead of second place. 7 Due to the overwhelming media coverage, they used the terms Al, machine learning, or deep learning to describe this technology. 3 Al includes machine/computer vision and natural language processing (NLP) agents that can perceive the environment and then react to it to obtain a specific goal. The basic idea is to “train” machines using algorithms and data so that they learn how to perform tasks to make inferences or predictions about how things will turn out. The industry tends to use two different groupings to illustrate the concept of machine learning, either by grouping algorithms according to learning scenarios or by grouping algorithms according to their form or function. 5 Deep learning is an advanced type of machine learning. DL methods are combinatorial non-linear models that automatically learn compelling features at multiple levels from high-dimensional, high-complexity raw data. 3 The term “depth” refers to the number of layers in the network; the more layers there are, the deeper the network. In its structure, modules at each level transform their input into higher-level, more abstract representations. 16 DL models can be considered end-to-end learning, where the learning process is not divided into modules or stages but is simply given training data in the form of “input-output” pairs that can be connected end-to-end to optimize the task. 12 The goal is to avoid the propagation of errors caused by traditional machine learning methods. 8
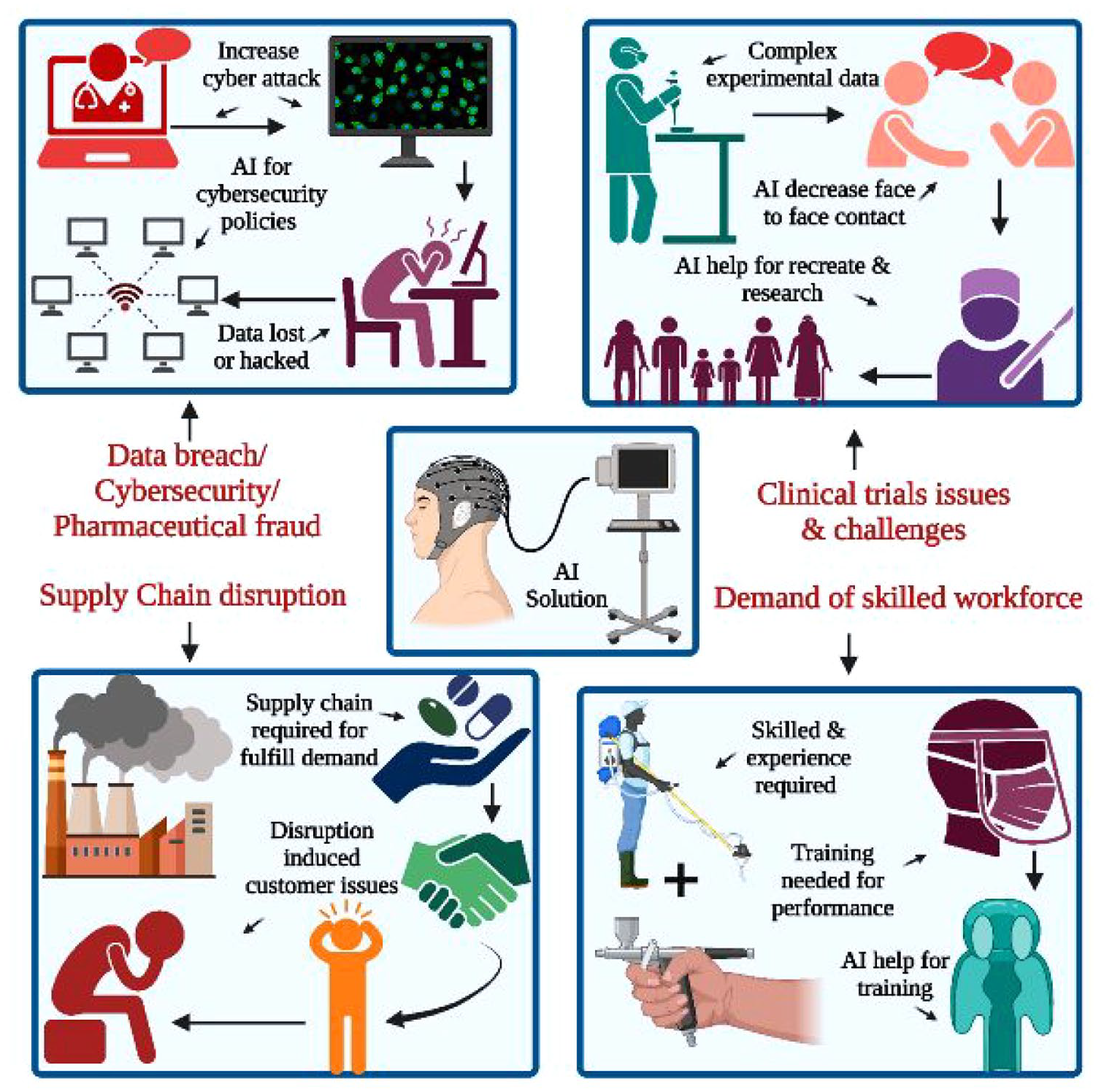
The data quality and algorithms implemented are crucial for AI pharmaceutical R&D enterprises. Currently, the limited volume of data for prominent molecule drugs is an essential constraint for AI pharmaceutical technology, leading to reduced accuracy of prediction models and difficulty in developing specific drug targets. Integrating dry and wet experimental data is the primary approach for supplementing data in AI pharmaceuticals. 17 Additionally, AI algorithms are often perceived as “black boxes,” lacking interpretability, which poses challenges in explaining the reasons behind algorithm outputs and ensuring the reproducibility of algorithm results. Balancing the requirements of data sharing and privacy protection is a problem that needs to be addressed. Lastly, AI pharmaceuticals require a lot of training data to support algorithm development, but data acquisition and quality issues remain challenging.
AI in Pharmaceutical Drug discovery has long been highly uncertain. 3 By removing some uncertainty from the drug discovery process, AI promises to improve significantly the chances of identifying new, commercially viable drug candidates, reducing costs and time. A predictive study in 2020 concluded that by investing heavily in AI, the pharmaceutical industry could increase its returns by more than 45%. 14 The drug development process, which aims to identify biologically active compounds to treat disease, begins with identifying molecular targets, then identifying active drug candidates and optimizing lead compounds for preclinical and clinical trials through to final regulatory approval. This process is time-consuming, high-risk, and expensive. The average cost of developing a new drug is between USD 100 million and USD 2 billion and can take between 10 and 17 years. Even if a drug candidate passes Phase I clinical trials, it has only a 5% chance of reaching the market. 3
Before 1980, the discovery of new drugs was achieved through random screening and empirical observation of the effects of natural products on known diseases. 5 This random screening process, although inefficient, resulted in several important drugs, such as the discovery of penicillin in the 1940s, which led to the effective control of once-incurable diseases such as tuberculosis and malignant bacterial infections 8 ; the discovery of antihypertensives drugs such as Prilosec, lipid-lowering drugs such as statins, and anticoagulants such as clopidogrel from the late 1970s, which led to the effective control of most cardiovascular diseases. After the 1980s, the drug discovery process was improved by high-throughput screening (HTS), which rapidly automates the screening of thousands of compounds against molecular targets or cellular assays. Identifying the immunosuppressant cyclosporine A in 1988 was a milestone in HTS. 15 Researchers continue to invest in new methods to improve the efficiency of the drug discovery process.
Generative AI in the pharmaceutical industry
Building upon the advancements in AI, generative AI (gen-AI) offers even more sophisticated capabilities, further enhancing the drug discovery and development processes (Figure 3). Gen-AI is transforming nearly all aspects of the pharmaceutical industry, revamping how companies operate and potentially unlocking billions of dollars in value. The McKinsey Global Institute has estimated that the technology could generate USD 60 billion to USD 110 billion a year in economic value for the pharma and medical-product industries, primarily because it can boost productivity by accelerating the process of identifying compounds for possible new drugs, speeding their development and approval, and improving the way they are marketed. 18

Generative AI flow of the method.
Pharmaceutical companies have long been practicing AI; researchers applied complex AI models to unlock disease mechanisms before last year’s explosion of interest. For example, AlphaFold2, ESMFold, 19 and MoLeR use deep learning to help predict the structures of nearly all known proteins, transforming our understanding of their underlying diseases.
The impending gen-AI-driven life-science revolution promises unquantifiable effects on human health and well-being. For example, an accelerated drug discovery process will help cure more diseases quickly, opening additional resources that could be applied to currently underserved areas. The ability to generate insights and patterns from vast quantities of patient data will spark more personalized treatments—and improved patient outcomes. Gen-AI tools could also make patient care more consistent by reducing deviations in the manufacture and delivery of therapeutics. Finally, by automating tedious and time-consuming tasks like document creation and record keeping, gen-AI can boost the productivity of researchers and medical liaisons so they can better serve clinicians and patients. As a result, gen-AI is expected to produce USD 60 billion to USD 110 billion in annual value across the pharmaceutical.
Before pharma companies can seize the opportunities gen-AI presents, they must step back and understand exactly what it can and cannot do—in other words, differentiate the reality of gen-AI from the hype that has come to surround it. For instance,
Gen-AI, on its own, cannot deliver the bulk of the value to be created. This is a disruptive moment for the entire field of AI, not for gen-AI alone. Traditional analytical AI models, such as those currently used to promote stakeholder engagement and help diagnose diseases, will continue to capture value. The difference is that new gen-AI applications will significantly enhance their capabilities.
Gen-AI cannot easily be plugged into existing data sets to unlock critical insights. Gen-AI cannot deliver results without a proper data architecture. Companies must build an intelligence layer to understand molecular structures, clinical operations, and patient data. A multipronged approach will be necessary to create a data infrastructure to run internal and external data sets. This is more than purely technical: data scientists must collaborate closely with leaders in business strategy, medical affairs, legal, and risk to set priorities and execute strategies.
Selecting the suitable large language model is not necessarily a critical strategic differentiator. Gen-AI models account for only about 15% of a typical project effort, and most of the work involves adapting models to a company’s internal knowledge base and use cases. That is particularly true in the pharmaceutical industry, given the complexity of its data and the uniqueness of its regulations and technology. To succeed with gen-AI, companies must integrate it across complex workflows to promote adoption and impact—a reality highlighting the need for effective change management.
Gen-AI will not instantly affect every part of the organization. As with any digital transformation, leaders must apply an end-to-end lens and prioritize only the use cases and applications that make sense for overall business goals. Those leaders must create a strategic road map to optimize the overall impact, the time to impact, and other important considerations. A “2 × 2 approach” is an effective strategy for companies getting started: begin with two use cases that require minimal disruption to the business, can build excitement across the organization, and have an impact most rapidly, as well as two other use cases that are potentially more transformational as longer-term goals.
Although the technology will affect all industries, it will have a powerful impact on pharmaceuticals. The reason is gen-AI’s truly multimodal nature. Foundational models are built not just on language but also on images, omics, patient information, and other types of data—and these are all required to explain and solve the processes of diseases and how best to treat compression—the decreasing amount of time they must capture a new drug’s value.
Future of gen-AI and drug discovery
Computer-aided drug design (CADD) has now improved the efficiency of new drug development. 11 CADD aims to use molecular modeling techniques to analyze the structure of numerous peptic and non-peptidic drugs quickly to study their interaction with pharmacological targets, their activity, toxicity, and bioavailability, allowing for better planning and guidance of drug discovery research. 10 For example, the antihypertensive drug captopril, the anti-HIV drugs saquinavir, ritonavir, and indinavir, the fibrinogen antagonist tirofiban, the glaucoma treatment doxorubicin, the antiviral drug zanamivir, the hypertensive drug aliskiren, and the protease inhibitor poprevir for hepatitis C—have all been discovered based on virtual screening techniques. Over the past decade, AI has been widely used in CADD to obtain more accurate predictive models, and the new AI-DD (AI in Drug Discovery) concept is gaining acceptance in the industry; future drug research and innovation will undoubtedly benefit from using AI in CADD. 20 For instance, gen-AI could help accelerate target identification, develop validation assays to test compounds, sign out of the most promising leads, and assist with preclinical testing to determine their effectiveness. Pharmaceutical manufacturers are already using foundational models for these purposes. In addition to natural language models such as BioGPT and Med-PaLM, the models that researchers use include image models to analyze microscopy and pathology data, chemistry models to improve predictions for functional readouts of small-molecule data, large-molecule models for protein folding and predictions, patient journey models to focus development efforts on promising indications, and multimodal models to combine these modalities and thus enable in silico experiments. Gen-AI is being implemented in the following ways.
Extracting scientific knowledge
Scientists spend much time extracting and summarizing information in documents such as patents, scientific publications, and trial data to better understand disease and drug targets. That is arduous and often provides incomplete or inaccurate information, given the sheer volume of data that must be processed. GPT-powered knowledge extraction—which uses AI algorithms to analyze unstructured data, including text, images, and other forms of information—can alleviate this burden. Unlike earlier solutions based on NLP, new gen-AI tools offer a much deeper and broader understanding of the medical context and intent. Researchers can, therefore, pose open-ended Q&As, quickly shift between different tasks, and frictionlessly integrate additional evidence through prompt engineering. Little to no further training is required to tailor information to specific use cases. This capability accelerates the research timeline and reduces the likelihood of errors caused by human oversight.
Additionally, ensuring the safety and security of sensitive medical data is paramount. AI tools must adhere to stringent data protection regulations and employ advanced encryption and access control measures to safeguard patient privacy and intellectual property. One example of these protection regulations is GDPR (General Data Protection Regulation), a landmark European Union regulation designed to give individuals control over their data and ensure it is handled securely and transparently. It applies to organizations worldwide that process the personal data of EU residents, enforcing strict rules to protect privacy. Another is HIPAA (Health Insurance Portability and Accountability Act), a US law established to safeguard sensitive health information, ensuring it remains private and secure while standardizing electronic health records (EHRs). It applies to healthcare providers, insurers, and related entities handling protected health information.
In silico compound screening
Drug development can be hindered by the difficulty of identifying and prioritizing the chemical compounds most likely to treat a particular disease successfully and, thus, most worthy of testing in laboratories. Gen-AI accelerates the screening process with state-of-the-art foundational chemistry models that map millions of known chemical compounds by their structure and function and overlay this information with known results for tested molecules. Like GPT-4, which is trained to predict the likely next word in a sentence, these models predict the next part (for instance, an atom) in the structure of a small or large molecule (such as an amino acid). The model learns fundamental significant and small-molecule chemistry principles through many iterations. This knowledge can then be used to train bespoke machine-learning models that offer still more precise predictions—even in largely unexplored areas of chemistry—that companies can prioritize for subsequent screening.
Optimizing strategy through indication selection
Gen-AI’s knowledge extraction capabilities can also help researchers determine which conditions or indications to target with a specific molecule—one of the most critical decisions facing biopharma companies. To make these calls, researchers must draw information from multiple sources, such as opinion leaders, literature reviews, omics analyses, trial data, and the activities of competitors. Yet, given the vastness of this information, indication selections often cover only part of the available evidence base, so conclusions may not be optimal. Gen-AI can help to address this issue by analyzing a wide range of structured and unstructured data sets. For example, real-world data (RWD)—drawn from doctor visits, insurance claims, electronic medical records, hospital data, and other sources—is often underused to select indications. With gen-AI, foundation models that treat medical events as words and patient medical histories as documents allow researchers to uncover the semantic similarity of different events, making it possible to estimate the biological proximity of one indication to another from a patient and clinical perspective. Moreover, information from molecular knowledge graphs can be tapped to reveal new connections (say, between entities such as proteins or human biological pathways) already identified in the literature or public data. These approaches can help uncover novel indications that can be rapidly validated through in vitro or animal models, increasing the likelihood of finding indications with a high probability of success and reducing the number of blind alleys (and their opportunity cost).
While gen-AI’s capabilities in analyzing RWD and molecular knowledge graphs hold immense potential, ensuring the safety and reliability of these tools is crucial. Current models rely heavily on the quality and bias of their training data, which can influence outcomes. To address this, robust validation protocols, algorithmic decision-making transparency, and continuous monitoring of inaccuracies are essential. Additionally, these tools must adhere to strict data privacy standards, such as GDPR or HIPAA, as mentioned previously, to protect sensitive patient information. As the technology evolves, further development is needed to enhance its interpretability, minimize bias, and ensure ethical application in medical research.
Higher possibility of success
About a 10% increase in the possibility of success for trials, about a 20% reduction in their cost and duration, and time to approval accelerated by 1–2 years—all leading to a potential double-digit impact on the net present value (NPV) of assets or portfolios.21,22 It takes, on average, 10 years and USD 1.4 billion in out-of-pocket costs to bring a single drug to market, and about 80% of those costs are associated with clinical development, according to researchers at the Tufts Center for the Study of Drug Development who reported a significant rise in the cost of drug development. 23 Clinical development is bringing therapies from lab to patient by rigorously testing a potential medication’s safety and efficacy in human subjects, a process characterized by lengthy clinical trial timelines and rigorous regulatory requirements. Gen-AI addresses these pain points by increasing efficiency across the entire clinical development process, unlocking economic value across three dimensions: up to 50% cost reductions enabled by the streamlining of clinical trial processes and auto-drafting trial documents; a 12-plus month acceleration in the time it takes to conduct a trial; and at least a 20% increase in NPV, thanks to enhanced health authority interactions, quality control, and improved signal management. 22 Across these three dimensions, we have identified four use cases with a strong potential for near-term impact.
Foundational approaches in drug discovery and development
Molecular characterization learning
A prerequisite for AI methods for drug molecule research is encoding molecules as fixed-length strings or vectors. 8 The vast chemical space of drug molecules often requires the selection of suitable molecular features that can accomplish the target task. Molecular characterization, also known as molecular descriptors, and selecting appropriate molecular representations are critical for accurately modeling and predicting small-molecule characteristics and biological activity. Chemical molecular characterization has essential applications in virtual drug screening, compound search/ordering, drug ADME/T prediction, inverse synthetic route planning, and other drug discovery processes. 24 Some of the molecular characterizations, when translated into computational space, can be presented in forms like
Simplified molecular-input line-entry system
SMILES (simplified molecular-input line-entry system) is one of the most used strings for drug representation, encoding molecular structures, and geometric properties. Due to its molecular linear representation, the SMILES string can be treated directly as text and is widely used as a molecular representation for deep learning models in a variety of drug design scenarios, in particular for inverse synthesis prediction models based on the seq-2-seq (sequence-to-sequence) method. 3 Another unique feature of SMILES is that by changing the atomic order, the same molecule can correspond to multiple SMILES, which can be used for data enhancement. 12
Molecular fingerprinting
A molecular fingerprint (MFP) is a string of bits encoding a molecule’s structural or pharmacological properties. 3 They are widely used for ligand-based similarity search and quantitative structure-activity relationship (QSAR) analysis in virtual screening (VS) of drugs; deep learning-based drug-target interaction prediction models also often use MFPs as input features. 3
Molecular fingerprinting based on the substructure, hash fingerprinting, and pharmacophore fingerprinting are the most common. The most representative substructure-based MFPs are the molecular access system and PubChem fingerprints for neighborhood and similarity searches; PubChemFP encodes 881 structural key types corresponding to the substructures of all compound fragments in the PubChem database. Hash MFPs such as Daylight FP, Morgan FP, and Extended Connectivity fingerprints (ECFPs) are commonly used for the similarity analysis of compounds. 13 Instead of using predefined substructures, hash fingerprints convert all possible fragments into values using a hash function. One of these, ECFP, is a recurrent fingerprint based on Morgan’s algorithm. It is often used as input to deep neural networks for bioactivity prediction and has shown good stability. 3 Pharmacophore fingerprints assign pharmacophore types to atoms in the chemical structure, generate multiple conformations, and construct binary fingerprints based on the resulting pharmacophore. The fingerprints are used as descriptors for partial least square QSAR models. By describing the aromaticity, hydrophobicity, charge, and hydrogen bond donor/acceptor of a molecule, the similarity between target binding sites can be assessed by considering a superposition of energy minimizing conformations of a set of molecules to extract pharmacophore features. 25
The two methods of molecule characterization described above provide many molecular descriptors. Still, the characterization results produced are limited by the domain-specific expertise of the computational chemist and depend on the algorithm used. 8 It is challenging to specify which structures and properties of molecules to characterize to obtain the desired results for subsequent downstream processing. 3
Graph neural networks (GNNs) offer the possibility of a complete and general approach to molecular characterization. By using graph nodes to represent atoms and graph edges to represent chemical bonds and then mapping their features onto a linear data structure in the form of a matrix or array, molecular graphs can be transformed from abstract mathematical concepts into concrete representations that can be processed on a computer. 3 In the molecular graph of a graphical neural network, each atom and bond has a corresponding initial feature vector as a feature matrix. The feature vector for an atom is the local chemical environment of the atom, including atomic type and formal charge number of hydrogens attached. The bond features can be the adjacency matrix, the type of bond, the shortest path, and the presence or absence of a particular ring. 25 GNNs can automatically learn task-specific molecular representations using graph convolution without the need for traditional manual molecular descriptors or MFPs and have a high degree of accuracy in predicting the properties of compounds. 26 Predicting a molecule’s chemical properties or activity directly from its structure has been a topic of great interest to the chemical community. 25 The graph neural network fingerprinting method, Neural FP, implements the use of graph convolutional neural networks to learn drug representations directly from molecular graphs; the weave model takes into account both atoms and chemical bonds in the molecular graph, further optimizing atomic features and atomic pair features, and demonstrates a high degree of accuracy in predicting water solubility, biological activity and toxicity of molecules, biological activity, and toxicity; Attentive FP is a graph neural network-based small-molecule representation framework that introduces a graph attention mechanism to construct node information that can learn local and non-local features of a given chemical structure, capturing fine substructures such as intramolecular hydrogen bonds and aromatic systems, thus providing excellent characterization learning capabilities for a wide range of different molecular properties. 13 In addition to drug property prediction and drug molecule characterization, GNN can also be used in ab initio drug design, interaction prediction, and inverse drug discovery. 12
Target discovery and validation
Targeted drug discovery is the mainstay of drug development. When a drug has a known target, it is easy to design drug screening experiments to discover therapeutics that act on the protein target. 27 By September 2021, of the 1619 drugs approved by the FDA, 1366 were small-molecule drugs, and 253 were prominent molecule drugs, involving 893 targets, of which 667 were human targets (the rest were pathogenic targets). 3 Failure to hit a target can lead to the waste of substantial R&D investments. For example, the clinical trials initiated by Pfizer, Roche, and Merck Sharp & Dohme for cholesteryl ester transfer protein inhibitors, a lipid-lowering target, all ended in disaster 5 ; the discovery of programmed death-1 (PD-1) has ushered in a new phase of biomolecule and tumor immunotherapy, and it is expected that there will be more than 20 PD-1 products on the market worldwide in the next 2–3 years.
On the other hand, even if a novel protein target with druggable properties is found, bringing a new chemical entity to market still faces an “absolute cliff” in terms of development time and cost, whereas discovering a new target or indication for a drug based on an existing disease can significantly reduce development costs. 28 The most famous drug redirection was sildenafil, and AI technology can make this serendipitous success tangible. 12 The combination of systems biology and AI algorithms to mine the correlation between multi-omics data and patient clinical health information, combined with NLP techniques to retrieve and analyze unstructured databases such as literature, patents, and clinical reports, can identify potential disease-relevant pathways, proteins, and mechanisms to discover new mechanisms and targets for drug development of novel chemical entities or drug redirection. 8
Systems biology approach
By studying the interrelationships and interactions between all components within individual biological systems at the molecular level (e.g., gene and protein networks associated with cell signaling, metabolic pathways, organelles, cells, physiological systems, and organisms), systems biology ultimately aims to build comprehensible models of whole systems and complete maps of organisms. 29 Network-based approaches infer new protein phenotypes or associations of protein functions by linking proteins/genes to different network pathways. 30 However, the high complexity of biological network interactions hinders the construction of network-based models for disease classification, personalized medicine, and prognosis. It often fails to provide stable pathway signatures of specific phenotypes or reliable disease biomarkers—data-driven, unbiased networks for identifying biomarkers of targets and diseases. 12 Using Bayesian AI analysis to combine molecular profiles from multi-omics data (genomics, proteomics, lipidomics, and metabolomics) with clinical health information to build causal inference networks, the difference between the “health” and “disease” network graphs can be used to identify disease drivers (targets and biomarkers), which were used to discover the novel tumor target BPM 42522, its lead molecule and its anticancer mechanism of action. 31
Combining knowledge mapping techniques with systems biology to build biomedical knowledge graphs has begun to play a critical role in medical practice and research. It helps to simplify complex biological systems and pathological processes, enabling researchers to understand better the principles involved; when combined with the context of a specific disease, biomedical knowledge graphs facilitate accelerated drug redirection and mechanical analysis of emerging human diseases such as COVID-19. 32 BenevolentAI has introduced JECS, a judgment-enhanced cognitive system that uses AI tools and biomedical knowledge graphs to identify potential drug candidates, enabling drug redirection by discovering new connections between large amounts of unstructured data such as disease, drug, and trial data and helping scientists find new indications for which known drugs may be applicable. 11 Similarly, MindRankAI is involved in building PharmKG39, a multi-relational attribute biomedical knowledge graph of drug-disease associations based on more than 500,000 relationships between genes, drugs, and diseases using a heterogeneous graph attention neural network containing 29 relationship categories and over 8000 ambiguous entities. Each entity in PharmKG is accompanied by heterogeneous domain-specific information extracted from multiple data sets, namely gene expression, chemical structure, and disease word embedding while preserving semantic and biomedical features. 33
Scientists at Insilico Medicine have developed the iPANDA (Insilico Pathway Activation Network Decomposition Analysis) method based on pathway activation analysis. 34 iPANDA is a powerful method for extracting biologically relevant features from large-scale transcriptomic and proteomic data. iPANDA uses gene expression data for biomarker identification. iPANDA takes as input the fold change between gene expression levels in tumor samples and the average expression levels of samples in the standard group and introduces gene importance factors to characterize the extent to which genes influence the pathway. However, the measure of gene centrality varies from algorithm to algorithm, and different algorithms can lead to highly variable results. 11 In this study, the degree of differential gene expression and the decomposition of pathway topology were integrated into a single network model, and statistical and topological weights were used to estimate gene importance.
Furthermore, gene modules reflecting gene co-expression were introduced, and the topological coefficients of each gene module were estimated to obtain gene co-expression data. Gene co-expression data were combined with gene importance factors to obtain pathway activation scores. Based on iPANDA, Insilico Medicine has established a new target discovery platform, PandaOmics, to develop new targets for IPF. They have identified and prioritized over 20 new targets by comparing histology data from fibrosis patients with that from healthy individuals to find significant differences between the two and by using iPANDA technology to find histology data on pathways that may affect these pathways. Subsequent screening for target safety and future value based on target knockout data has led to the identification of novel targets for treating IPF, and the project is now rapidly progressing to the clinical stage. 25
Target structure-based approaches
Confirmation of novel targets in drug development (i.e., target selection or prioritization) is still an uncertain process, and it is essential to accurately map the interactions between approved drugs and their efficacy targets (i.e., the targets on which the drug exerts its therapeutic effect). 8 Structure-based computational approaches to target discovery can be used as a strategy to complement experimental approaches such as reverse docking, pharmacophore, binding site similarity, and fingerprint-based interactions. 35 Among these, reverse docking has become one of the most effective tools for identifying potential targets for a given compound, not only for target validation but also for predicting toxicity and adverse side effects and for discovering unknown novel targets for drugs or natural compounds. 3 Potential targets for tea polyphenols and ginsenosides were identified using the PDTD (Potential Drug Target Database), a large target protein database for reverse docking screening. The target structure dataset limits the reverse docking approach; the PDTD, released in 2008, contains approximately 1100 protein entries with 3D structures, and its data is extracted from the literature and several online databases (e.g., TTD, DrugBank, and Thomson Pharma) and includes information on 830 known or potential drug targets. 36 Only about 11% of the human proteome has been annotated with small-molecule probes, and the function and role of a third of the proteins in human biology and disease are not yet known.
Three methods, Nuclear Magnetic Resonance (NMR), X-ray crystallography, and cryoelectronic microscopy, are now widely used for the structural resolution of proteins and have provided much information about the structure of proteins and drug receptors, and many drugs, such as angiotensin-converting enzyme inhibitors, have entered clinical practice based on structural information.37–39 Cryoelectron microscopy equipment costs around USD 20–60 million, and the synchrotron light source required for crystallography costs hundreds of millions to build. 40 Experimental methods to decipher a protein structure can take anywhere from a few weeks and months to several years, depending on factors such as sample availability and protein complexity. 41 Therefore, AI protein structure prediction algorithms will be an essential adjunct to protein information-based target validation methods. AlphaFold2 achieved a median global distance test (GDT) score of 92.4 across all targets, close to the quality provided by gold-standard experimental techniques such as X-ray crystallography. 8 AlphaFold2, developed by DeepMind, uses a DNN (deep neural network) architecture trained on 170,000 protein structures from the PDB (Protein Data Bank) to predict the distribution of distances between pairs of amino acids and the torsion angles between the chemical bonds connecting these amino acids in proteins. 42 The methods and architecture behind AlphaFold2 have recently been published, and, in collaboration with EMBL-EBI (European Molecular Biology Laboratory-European Bioinformatics Institute), AlphaFold2 predicted 3D structures providing structural coverage of 98.5% of the human proteome have been made freely available to the scientific community. 5 Although AlphaFold3 does not yet provide a good description of the side chain structure and dynamics of proteins, and it is difficult to predict the structures of multi-structured domain proteins, multimeric protein complexes, and membrane proteins, the AlphaFold3 protein structure library provides us with a library of reverse docking structures that almost covers the human proteome, providing a list of potential target proteins for further studies and helping to address the limitations of target structure data sets in reverse docking, reverse docking has the potential to become a handy tool for drug discovery and thus advance drug discovery. 43
Small-molecule drug discovery
In recent years, deep learning has significantly impacted fields such as image analysis and NLP. Inspired by these successes, computational chemists increasingly use generative models to generate new molecules and predict their properties. 32 The chemical space contains approximately 1060 to 10,100 possible small molecules. Drug discovery efforts need to find molecules that meet multiple criteria, such as biological activity, metabolic stability, and potency-like finding a needle in a haystack. As a result, only a tiny fraction of the theoretically possible chemical space can be explored in wet experimental studies. Computer modeling techniques can further enhance the biological screening of large compound sets and the design of synthetic routes to complementary compounds, and they are an essential component of early drug discovery. 5
Molecular generator techniques
One of the keys to compound design and predictive modeling is the choice of molecular representations. On the one hand, text or string encoding of molecules is computationally inexpensive and commonly used data structure in molecular generators. For generative modeling, SMILES-based string encoding typically generates a token for each atom, then converted to a “one-hot” string representation. 44 A generative model using one-hot string representations generates a distribution of each token, which is then sampled to generate a new structure for the SMILES encoding. On the other hand, graph-based generative modeling is an emerging area of research, for example, using graph convolutional policy networks or deep learning to generate molecular structures. Rule-based graph generative models often produce formally correct structures but are computationally expensive. The combination of flexible neural network architectures and various molecular representations has led to various architectures and solutions for molecular generative models. 45
Synthetic route planning
The use of computer-aided synthetic planning (CASP) can be traced back to the pioneering work of E. J. Corey, who formalized the concept of “inverse synthetic analysis” in the late 1960s. 46 CASP incorporates the ideas of inverse synthetic analysis to help synthetic organic chemists select the most efficient and cost-effective synthetic routes. It can be used to predict selectivity and by-products and to suggest and evaluate reaction conditions. Over the decades, computational methods have evolved from expert systems based on hand-coded reaction rules and templates to data-driven AI-assisted synthesis planning. 30 Some AI algorithms are now available to recommend feasible synthetic routes for various reactions: with or without reaction templates, operating at the mechanical or global reaction level, and using molecules represented as fingerprints, graphs, or SMILES strings. CASP can help chemists make better decisions, increasing efficiency and productivity by reducing synthetic failures and accelerating the drug discovery cycle’s DMTA (Design-Make-Test-Assess) phase. 3
Rule-based approaches use expertly coded rules and heuristics extracted from reaction databases and literature to suggest synthetic routes, often called “template methods.” In a rule-based approach, reaction rules are manually extracted and coded, which is limited by the inability to expand with the exponential growth of the chemical literature and by the fact that its knowledge base is limited and cannot be fully covered. Synthia (Chematica) is an inverse synthesis software that uses a library of expertly coded rules for chemical synthesis planning. 5 To overcome the limitations of the rule base, Synthia uses computational methods to automate the extraction of reaction rules from reaction datasets. Its template extraction algorithm is based on Ambit-SMIRKS, defined explicitly for describing chemical reactions. It has collected an expert-coded reaction rule base of approximately 50,000 rules over 15 years. 10 Synthia’s core algorithm is a decision tree in which various conditions specify the range of possible substituents or atom types; a scoring function and dynamic planning algorithm are then used to construct complete synthetic pathways, making decisions for each inverse synthetic step, allowing synthetic routes to be proposed for all targets in 15–20 min. In 2016, Szymkuc et al. used Synthia to design synthetic pathways for eight structurally diverse and synthetically challenging target molecules, marking the first successful use of synthetic planning software to guide multi-step synthetic routes. 40 They selected the highest-scoring synthetic route to synthesize the targets, achieving up to 98% yields.
Interestingly, the synthetic route proposed by Synthia differs significantly from the original synthetic route disclosed in the patent, providing higher yields with fewer synthetic steps. 36 AI techniques have also been used in recent years to extract reaction rules. Segler et al. pioneered using a neural-symbolic approach to extract inverse synthesis rules from the Reaxys database autonomously, without expert input. 47 These rules were combined with modern Monte Carlo tree search algorithms for reaction prediction to select the most promising inverse synthesis routes. However, using templates brings disadvantages such as high computational costs and incomplete rule coverage, limiting scalability. 48
Given the shortcomings of the template approach described above, the template-free approach draws inspiration from NLP and treats forward or inverse synthetic prediction as a Seq2Seq mapping problem. 10 Since molecules can be represented as SMILES strings, each chemical reaction can be encoded as a sentence and treated as a chemical language translation problem. 11 The first template-free approach to inverse synthesis analysis is based on the Seq2Seq model, which is entirely data-driven, trained end-to-end on a subset of experimental reactions with labeled reaction types, and consists of a bidirectional LSTM (long short-term memory) encoder and decoder with an additional attention mechanism that maps the SMILES of the reactant representation to the SMILES of the product representation. 49 The method’s performance is comparable to that of a baseline model of a rule-based expert system.
The small-molecule drug design and optimization
Structure-based virtual screening (SBVS), also known as target-based virtual screening (TBVS), is a robust, effective, and promising CADD technique. 45 The SBVS approach predicts the interaction of a target protein with many compounds from a database based on its 3D structure and then scores. It ranks the compounds according to their affinity for the target receptor binding site, thereby identifying compounds that are more likely to be of interest to the molecular target. This leads to identifying ligands that are more likely to be pharmacologically active against the molecular target. 25
The SBVS technique, molecular docking, which explores the geometric fit between ligand and target, attracted attention for its low computational cost and good performance as soon as it appeared in the 1980s and became widely used in the 1990s as computational power increased and structural data on target molecules accumulated time and cost; the presence of solid molecules is not required; and computational testing can be performed before molecular synthesis. However, existing SBVS are often system-specific and ineffective in more general situations; the high complexity of ligand-receptor binding interactions makes it difficult to parameterize them to accurately predict the correct binding site and classification of compounds, resulting in high false-positive and false-negative rates for SBVS. 12 Docking protocols are essential to achieve accurate SBVS and consist of two main components: a search algorithm and a scoring function. The search algorithm, which systematically searches for ligand orientation and conformation at the binding site, and the scoring function, which predicts the binding affinity between the target and its candidate ligands, are critical to the success of docking. 40
In general, scoring functions have three important applications in molecular docking
to determine the binding/alteration sites of targets and ligands as well as the binding confirmation
to predict the binding affinity between proteins and ligands
to optimize potential ligands. 32
There are three main types of traditional scoring functions, namely force field-based, empirical, and knowledge-based scoring functions, with machine learning-based scoring functions, which have emerged in recent years, being considered as a fourth type. 50
Traditional scoring functions have well-known limitations—they do not adequately consider conformational entropy (the flexibility of the protein) and solvation energy. 51 Based on a large amount of experimental data available, AI algorithms can build non-predefined scoring functions that are data-driven by implicitly learning the eigenvectors of protein-ligand binding and their non-linear relationship with affinity. Many researchers have used machine learning scoring functions to improve SBVS algorithms, such as RF-Score-VS and SFCscoreRF based on RF (random forest), SVR-KB/-EP and ID-Score based on SVM (support vector machine), and NNScore 2.0 and CScore based on early artificial neural networks. 52
While traditional machine learning approaches still rely on expert knowledge and feature engineering, the rise of deep learning algorithms offers a new direction for scoring function modeling. 16 CNNs (convolutional neural networks) can automatically extract features directly from 2D or 3D structures to predict the binding affinity of proteins to ligands. The 3D lattices of protein-ligand structures generated by docking can be used as input to CNN models, from which relevant features such as complex atom types, partial atomic charges, and distances between atoms are automatically learned and extracted to build regression models for predicting affinity or classification models for predicting binding or non-binding, with better predictive performance than other docking methods. 12 Deep learning techniques, particularly CNNs, have breathed new life into SBVS. Classical scoring function approaches use predefined theories to design functions based on linear relationships, and the introduction of AI techniques can implicitly capture intermolecular binding interactions that are difficult to model explicitly. Although scoring functions generated by deep learning techniques may not always be more predictive than established machine learning methods, and further optimization of training efficiency and interpretability is required, existing docking tools continue to see practical improvements by introducing deep learning. SBVS will be one of the most promising techniques in the drug discovery process in the coming years. 45
Ligand-based virtual screening (LBVS) assumes structurally similar compounds have similar biological activities. QSAR, pharmacophore, and structural similarity matching have long been the most used LBVS methods. 47 The QSAR model has been developed over half a century as one of the significant computational molecular modeling approaches and aims to find a mathematical relationship between the molecular properties of a compound (e.g., polarity, lipophilicity, electrical and spatial properties or specific structural features) and certain activity indicators (affinity to receptor sites, inhibition constants, rate constants, etc.). As a modification of QSAR, 3D-QSAR calculates binding affinity by reading parameters directly from the 3D structure of the compound. Comparative molecular field analysis (CoMFA) is an essential method for 3D conformational analysis. The 3D pharmacophore is a conformational analysis and molecular stacking of known active compounds to obtain information about the moieties that play a key role in their activity. In pharmacophore-based VS, 3D pharmacophores developed from a set of active ligands, target-ligand complexes, or protein targets are screened against a virtual library of molecules, from which molecules that meet the requirements of the query pharmacophore are retrieved. 53 VS models based on AI algorithms take as input descriptors based on the physicochemical properties of molecules and/or fingerprints based on the topology to build regression or classification models of activity, providing a more flexible approach to LBVS that is no longer dependent on program-specific functionality. Bayesian algorithms, SVM, RF, and artificial neural networks have been widely used to build QSAR models and have provided many compelling applications in LBVS. 3 DNNs have demonstrated superior predictive performance compared to machine learning methods such as Bayesian, RF, and SVM, and multi-task DNNs have shown further performance improvements, with multi-task DNNs on 200 different targets emerging for large-scale applications. A QSAR model built using multi-task DNNs for 15 different tasks won the 2020 Kaggle challenge sponsored by Merck Sharp & Dohme. 54 DeepTox, a multi-task DNN-based toxicity prediction method, won the 2014 Tox21 dataset challenge, which required the prediction of compound toxicity using a dataset of 12 high-throughput toxicity assays for 12,000 compounds. 8 Schrödinger, Inc. has integrated its AutoQSAR with DeepChem (https://deepchem.io/), making it easy for non-computing experts to build and apply high-performance deep learning QSAR models on large datasets. 55 Recently, molecular prediction models for antimicrobial activity based on the MPNN (message passing neural network) model have identified eight antimicrobial molecules with structures different from conventional antibiotics from a large database of over 107 million molecules and identified a novel antibiotic, halicin, which inhibits the growth of Escherichia coli. 5
The combination of the pharmacophore concept with AI techniques is still in its infancy, and future research will focus on using pharmacophore features as molecular descriptors for AI models or using AI methods to generate pharmacophores from large amounts of data. For example, when Pharm-IF, a pharmacophore-based interaction fingerprint, was used as input to several machine learning algorithms to rank small-molecule docking poses, the models combined with SVM showed the best enrichment rates over other machine learning algorithms and docking scoring. 56 It is believed that AI algorithms combined with increasingly sophisticated molecular characterization methods will soon become the dominant LBVS technique.
Advances in ADMET prediction for drug development
ADMET is a crucial indicator for assessing whether a small-molecule compound can become a drug, covering pharmacokinetic and toxicological issues such as whether the drug can be effectively absorbed into the body and reach the target tissue. It covers pharmacokinetic and toxicological issues, such as whether the drug is effectively absorbed into the body and reaches the target tissue. Many clinical trial failures have been attributed to deficiencies in the ADMET properties of drug candidates. Conducting ADMET property evaluation studies at an early stage of drug development can effectively address the safety and efficacy issues of drug candidates and improve the success rate of drug development. 29 However, the experimental methods used for ADMET property evaluation are expensive and time-consuming, limiting the understanding of early compounds and affecting further biological validation. With the development of computer technology and cheminformatics and the accumulation of experimental drug data, ADMET prediction models represented by machine learning and deep learning can learn the association between chemical structures and pharmacokinetics from ADMET data, preventing medicinal chemists from exploring the poor and unknown chemical space and thus finding the best molecules. 57
It can be argued that ADMET prediction is essential to drug discovery and development. Major companies and institutions at home and abroad are committed to combining conventional wet experiments with computer experiments to help drug developers analyze the ADMET profile of compounds from a computational perspective, resulting in several computer-aided ADMET software, databases, and online services. 8 For example, the QikProp module of Schrödinger software can predict the log P, log S, Caco-2 cell permeability, serum protein binding activity, hERG-Kion channel blocking, etc. 51 GastroPlus has been widely used by the FDA, NMPA (National Medical Products Administration), and other national drug regulatory authorities. It can predict pharmacokinetic parameters such as physicochemical properties, absorption, distribution, and metabolism, as well as the in vivo course of the drug after ocular and pulmonary administration. 11 The SwissADME molecular modeling team, developed by the Swiss Bioinformatics Institute, can calculate physicochemical descriptors to predict pharmacokinetic properties such as oral bioavailability, blood-brain barrier permeability, and the potential of compounds to bind to metabolic enzymes, drug formation, and drug chemistry friendliness. They have also independently developed a range of online prediction tools for ADMETs that are widely used by researchers in the United Kingdom and abroad. 46 ADMETlab uses molecular fingerprinting features such as MACCS and ECFP4 to train machine learning models such as RF, SVM, and Plain Bayes for classification and regression prediction of multiple ADMET properties; ADMET SAR also uses MACCS to construct MFPs to train machine learning models such as SVM and achieves better prediction performance in 22 classification tasks, which DrugBank, a drug database, has adopted. Currently, machine learning-based prediction tools are the most widely used, but the use of MFPs and molecular descriptors as features can cause a significant loss of molecular structure information and may limit the prediction performance of the models. 58
Deep learning-based ADMET prediction methods automatically extract feature representations of the input to fit more complex associations. As reported in the 2020 Kaggle competition, DNNs improved performance by an average of 10% over RF models on 15 large analytical datasets. 59 Researchers at major pharmaceutical companies Vertex Inc., Eli Lilly & Co, and Bayer AG found DNNs comparable to or slightly better than mainstream machine learning models when trained on their large private ADMET datasets.60–64 The recent emergence of GNNs has added a new dimension to the design space of ADMET models. These graph neural networks use graph structures to represent molecules and, through data-driven training, convert molecular structure information into continuous low-dimensional dense vectors, an information representation superior to the use of high-dimensional sparse MFPs. 65 The superiority of graph neural network models for predicting drug properties has been demonstrated by models such as Molecule-Net and Chemi-Net. Chemi-Net, an entirely data-driven, domain-knowledge-free deep graph convolutional network approach developed in collaboration with Amgen, was compared with Amgen’s Cubist machine learning program for large-scale ADME property prediction. The results showed that for all 13 datasets, Chemi-Net was significantly more accurate in ADME prediction than the Cubist benchmark, helping to accelerate drug discovery. 28 In addition, graph neural networks can use built-in interpretable methods such as SAMPN, a message-passing neural network based on a self-attentive mechanism. Self attention-based message passing network (SAMPN) outperforms both graph neural networks and RF without adding an attention mechanism in predicting lipophilicity and water solubility, and it can visualize the contribution of each atom to the predicted properties through the attention coefficient. 66
Currently, the application of machine learning in predicting ADMET properties is still constrained by the compounds available in publicly disclosed training data. Despite the successful application of some AI models in ADMET and activity prediction, a key challenge is the availability of data and the generalizability of data-dependent models. Furthermore, the methods mentioned above often only measure the similarity of physicochemical properties between approved drugs without fully considering the characteristics of drugs in biological systems (such as permeability and clearance rate). Therefore, a single score cannot fully encompass the complex property space of drugs, significantly limiting the guidance for compound optimization.
Accelerating clinical trials
Despite promising advances in systems biology and a significant increase in the availability of high-throughput biological data, the pharmaceutical industry is experiencing a decline in R&D efficiency, with clinical trial failure rates of up to 95% in oncology and other disease areas. High clinical trial failure rates lead to high costs and inefficiencies in drug development: bringing an entirely new chemical entity to market can take up to 7–10 years of clinical trials at a capitalized cost of USD 1.46–2.56 billion. Losses per failed clinical trial range from USD 800 million to USD 1.4 billion, including the investment in the trial itself and the loss of preclinical development costs. Applying AI technology to critical steps in the design of clinical trials can help achieve more accurate patient stratification and improve recruitment efficiency, thereby increasing the success rate of clinical trials.67–70
Over 75% of the cost of developing new chemical entities is spent on in vivo studies, which means that improvements in calculation methods made early in drug development have a limited impact on overall development costs. Phase III clinical trials would involve testing large numbers of patients, and the financial cost of failure at this stage would be catastrophic. Ideally, AI models should be used to predict late-stage trial outcomes. In silico clinical trials (ISCT) could significantly reduce clinical trial costs while increasing overall success rates. 71 In 2005, a white paper first described virtual physiological human, which aims to develop patient-specific computer models to support clinical decision-making and models to form virtual patient groups to test the safety and efficacy of new drugs and medical devices. 72 In addition, it is conceivable that a virtual patient group could complement clinical trials (reducing the number of patients enrolled and increasing statistical power) and suggest clinical decisions. ISCT typically integrates physiological and pathological information about patients at different spatial and temporal scales, aiming to generate patient-specific predictions and treatment plans for diagnosis, prognosis, dose selection, or specific patient groups. 25 However, using ISCT to reduce, improve, or partially replace in vivo experiments remains challenging.
On the one hand, Big Pharma and research institutions must continually address the inherent complexities associated with accurate, quantitative modeling of organisms; otherwise, clinical trials alone will not provide sufficient information on structural and design properties to explain the failure of drug candidates, and their reliability remains to be proven. 5 As a result, current AI technologies are primarily designed to improve clinical success by intervening in several critical aspects of clinical trials. Linking patient genetic characterization data, EHRs, medical literature, and clinical trial databases can predict clinical toxicity and trial success, improve trial design, assist with patient-trial matching and recruitment, and monitor patients during trials to improve patient adherence. 25
Prediction of clinical trial results
Deep learning models based on the analysis of drug response and side effects to predict the outcome of phase I/II clinical trials can significantly improve the success rate of clinical trials and help to improve the drug development process. 8 With many clinical trials likely to fail due to toxicity, ProCTOR uses a combination of chemical features of drugs and target-based features to model the distinction between FDA-approved drugs and FTT (failed for toxicity in trials) drugs. The method uses a 48-feature set of 10 molecular features, 34 target-related features (e.g., target tissue selectivity), and four drug-like rules to construct an RF classifier that directly predicts the likelihood of a drug being toxic in clinical trials. 73 In addition to toxicity, more than two-thirds of clinical trials fail for other reasons, including efficacy, strategic, and financial reasons. Efficacy is a very complex issue, and combining in vitro cellular models with data on drug side effects can directly predict the success or failure of clinical trials. 74 Insilico Medicine built a deep neural network based on pathway analysis techniques to predict the side effects of compounds based on the transcriptional changes they cause in cell lines and the success or failure of the associated clinical trials. The study thoroughly analyzed transcriptomic data from drug-induced perturbations in cell culture and used pathway activation scores estimated by the iPANDA algorithm as input to build neural networks to predict clinical trial outcomes for each of the 46 side effects. 8
Clinical trial design
Using AI technology to support clinical trial design and enable efficient patient stratification, recruitment, and monitoring can help improve the efficiency and success of clinical trials. In collaboration with Johns Hopkins University and the National Cancer Institute, rational clinical trial design presented the iPANDA pathway analysis algorithm for head and neck squamous cell carcinoma (HNSCC). iPANDA was applied to transcriptomic data from 359 oral squamous cell carcinomas (OSCC) and 86 white spot samples (precancerous lesions) to identify differentially dysregulated pathways between these tumors and normal oral mucosal tissue and to elucidate the signaling pathways from white spots to OSCC. 25 This work will contribute to a better understanding of the complex signaling networks behind HNSCC and may help to develop new preventive, diagnostic, and therapeutic approaches. Furthermore, with the advent of immune checkpoint inhibitors for the treatment of HNSCC, there is a need for more reliable characterization of the tumor microenvironment, signaling pathways, and genetic alterations associated with CD8+ T-cell infiltration in HNSCC. The study used RNA sequencing, 10 chemokine signatures to classify HNSCC patients into high and low CD8+ T-cell infiltration subgroups (Tc-cell inflammed phenotype-H ( TCIP-H) and Tc-cell inflammed phenotype-L (TCIP-L), respectively), and iPANDA analysis to analyze differences in signaling pathways, somatic mutations, and copy number aberrations between TCIP-H and TCIP-L tumors. The study found that TCIP-H HNSCC tumors are rich in multiple immune checkpoints and are promising candidates for potential combination immunotherapy, providing a rationale for designing rational combination immunotherapies. 3
Extraction of electronic medical records using NLP techniques can be used to match patients to clinical trials. For example, IBM Watson has developed a clinical trial matching system that uses structured and unstructured data from patients’ electronic medical records and available trials to create detailed patient clinical profiles. Comparing a patient’s clinical profile with the required clinical trial eligibility criteria can help optimize clinical trial protocols for eligible patients or find patients who meet the requirements for a particular trial. 75
However, limited data from clinical trials and electronic medical records are insufficient to describe the intrinsic complexity of organisms, and the lack of interpretability casts doubt on the reliability of protocols and poses some ethical risks. The further introduction of systems biology approaches based on medical data is now the dominant research direction, with systems biology helping to provide biological insights into the mechanism of action of drug candidates. For example, Insilico Medicine’s iPANDA used a microarray analysis quality control (MAQC) dataset based on multiple sources of paclitaxel-based neoadjuvant breast cancer therapy to identify a highly robust set of biologically relevant pathway features that were successfully used to characterize breast cancer patients according to their sensitivity to neoadjuvant treatment. GNS Healthcare’s REFS (Reverse Engineering and Forward Simulation) machine learning platform combines longitudinal electronic medical records, pharmacy and medical claims, next-generation sequencing, and other medical treatments. GNS Healthcare’s machine learning platform, REFS, combines patient data such as longitudinal electronic medical records, pharmacy and medical claims, next-generation sequencing, and other “histological data” into computational models and applies machine learning techniques to answer complex healthcare questions, to uncover hidden drivers of cancer progression and drug response at the patient level, thereby identify new targets and biomarkers of disease, which can then be used to stratify patient populations 76 more accurately.
Traditional adherence measures, such as pill counts and other self-reported data, are susceptible to patient manipulation and data distortion. Abbvie uses the facial and image recognition algorithms of the AiCure mobile SaaS platform to monitor users by having patients record a video of themselves swallowing pills with their smartphones, and the AI platform verifies that the right person has swallowed the right pills. Adherence increased from 50% to 90% in the group of patients with schizophrenia within 6 months. 77
Drug repositioning: Unlocking new therapeutic potential
Bringing new chemical entities to market faces an “absolute cliff” regarding development time and cost; discovering new indications for a drug based on an existing drug for one disease and using it for another can significantly reduce development costs. Drug repositioning, or drug repurposing, is a drug discovery strategy to identify new therapeutic effects of an approved or candidate drug outside its original therapeutic use. 78 This allows retargeted drugs to rush into Phase II and Phase III clinical studies, and the associated development costs can be significantly reduced, given that the drug’s pharmacokinetic, pharmacodynamic, and toxicity profiles have already been established in the initial preclinical and Phase I studies. Traditionally, success stories of drug repositioning have come primarily from an understanding of drug pharmacology or retrospective analysis of the clinical effects of a drug. For example, sildenafil citrate was originally developed as an antihypertensive drug but was later repositioned by Pfizer for the treatment of erectile dysfunction based on retrospective clinical experience; the use of thalidomide for the treatment of erythema nodosum leprosum and multiple myeloma was based on serendipitous discovery. 28 Rapidly evolving AI drug design methods can make such serendipitous successes traceable.
Using a combination of systems biology and NLP techniques, the study of patients’ off-label drug use is a retargeting approach favored by pharmaceutical giants, using large-scale histological data and patients’ EHRs. 3 BenevolentAI’s judgment-enhancing cognitive system, JACS, uses AI tools and biomedical knowledge graphs to identify potential drug candidates by discovering new connections between large amounts of unstructured data, such as disease, drug, and trial data, to enable drug redirection and help scientists discover more valuable indications for drugs. Johnson & Johnson has entered into a collaboration with BenevolentAI and signed an exclusive licensing agreement for some clinical-stage drug candidates to redevelop Bavisant, a highly selective histamine H3 receptor inverse agonist previously developed by Johnson & Johnson for failed attention deficit hyperactivity disorder, for extreme daytime sleepiness in patients with Parkinson’s disease and has opened Phase II clinics. 79 In February 2020, shortly after the World Health Organization declared the COVID-19 outbreak a public health emergency of international concern, BenevolentAI used knowledge mapping to identify Baricitinib, a drug developed by Elli Lily for the treatment of rheumatoid arthritis, as a potentially effective COVID-19 drug within days, TwoXAR used its DUMA™ drug discovery platform to mine the relationships between multi-omic data, protein interactions, chemical structures, and patient clinical data for new uses of old drugs and found that exenatide and olopatadine were more effective in animal models of rheumatoid arthritis. 80
In summary, AI technology has generated numerous typical application cases in the pharmaceutical field. In drug development, AI can learn and comprehend patterns and rules of chemical reactions, efficiently identify targets, and design and screen candidate molecules, providing strategies and pathways for drug synthesis and predicting drug kinetics and adverse reactions, thereby shortening the drug development cycle. 81 However, AI faces technical challenges in predicting adverse drug reactions and interactions, such as low data quality and prediction accuracy. Moreover, drugs solely generated by AI technology have not yet been marketed; thus, the effectiveness of AI applications in the development field remains to be tested in the market.
Application Challenges of AI in the pharmaceutical industry
We have long believed that introducing new technologies, such as the digitization of drug discovery and development and AI, will be an irreversible trend. According to the above analysis, we must acknowledge that the difficulty of accessing the various elements of resources varies greatly and that the maturity of AI macromolecule drug development in each segment will be pulled apart. Based on the above judgment, we believe that at this extremely uneven data distribution stage and immature data sharing models, current AI macromolecular drug discovery companies must build their data asset production capabilities to establish true differentiation. The latitude of data production could be antibody screening (single B-cell analysis), target protein-function relationships (proteomics), target epitope structures, peptide structures, etc. The data production platform must be unique and closely related to drug development.
Regarding data sharing, federated learning will be easier to implement on the ground than participating in or building a DAO (decentralized autonomous organization). Forming a federation will focus more on the standardization of data and the degree of digitalization of the companies involved, and flexible cooperation between two or three companies will be easier to implement on the ground. The barriers formed at the data level through the volume of data or the way data is created will be more reliable.
Based on current observations, it is relatively challenging to protect the property rights of algorithms, and therefore, the construction of barriers is relatively short-lived. Based on the perspective of combining wet and dry experiments, AI macromolecule drug development companies still need some innovative algorithm development capabilities, which are concentrated on the application side. Innovation on the application side of the algorithm should be understood as development needs to be systematic and continuously iteratively updated. The output should make it as easy as possible for life scientists to understand and smoothly interface with wet experiments (e.g., whether the data predicted by the algorithm has realistic meaning, whether it is interpretable, whether it can give corrections based on predictions, etc.). In the algorithm and arithmetic part, we support companies collaborating more and polishing their capabilities with an open attitude.
In terms of business model, no matter what path is chosen, it needs to be based on the company’s deep understanding of drug development; otherwise, the CRO (contract research organization) business will not be able to standardize its business and will be limited in expanding its categories and increasing the number of customers. In the drug development business, wet experiments (accumulated over several months) take up much more time than dry experiments (accumulated over several days), and the team’s lack of understanding of drug development will ultimately dilute the efficiency gains of dry experiments by the inefficiency of wet experiments. From the current development of public companies, it is easier to realize the value of a company’s direct involvement in drug development than CRO services. The core reason is that downstream customers lack evaluation criteria for the quality of AI algorithms as CRO services and have a low willingness to pay, a particularly evident feature in China. Drug development companies need at least two dimensions of life sciences and algorithms talent pool and, more importantly, develop their methodology to string wet and dry experiments (i.e., choose what kind of data set to stock, what link model training has good prediction results after, combined with prediction results how the team decides to advance the project), to achieve achievements beyond traditional drug development, which is AI macromolecule drug development companies A sufficient condition for success. To achieve the above, the company needs to master technologies that may not only be strictly defined as machine learning algorithms and biopharmaceuticals but also incorporate knowledge from fields such as synthetic biology (solving the synthesis of artificial proteins) and engineering automation (digital adaptation of laboratory automation).
Because of the current challenges, pharmaceutical companies have a huge demand for advanced technologies to facilitate discovering and validating new drugs. There have been hundreds of collaborations between pharmaceutical and AI technology companies worldwide. It is fair to say that the traditional pharmaceutical industry is experiencing a shift from skepticism to interest in AI. But how far is it from interest to trust? The use of AI in pharma is expected to bring the entire AI ecosystem into pharma. So, will computational pharma and traditional pharma become parallel models in the future, like online and offline (online shopping is essentially VS)? That remains to be seen. Can the myriad variables of a complex biological system be quantified and analyzed accurately enough to identify new drug targets and better assess the effects of drugs? There are still many unknowns to be explored. However, whether AI can reshape and transform the drug discovery process, extracting value from all data across the drug discovery cycle is the way forward. Data is not the same as science, but almost all scientific advances are identified and determined from data. As the volume of data continues to grow, drug development data is evolving into big data. And AI is currently the most ideal and effective way to handle big data.
Conclusion
The pharmaceutical industry has enormous and growing amounts of data, and in terms of models, the best AI pharma model is not to build pure AI processes. Combining humans and AI is often superior to human processes or AI processes alone. Just as in chess, the combination of a human and a computer algorithm can usually beat a human or a computer algorithm alone. AI technology methods need to be sorted out and developed. AI’s attention, exploration, and application trials in all sectors of society will inevitably accelerate the maturation and innovation of AI technology methods. When the logic of the “large data → more accurate models → better drugs → more and better data” cycle matures in practice, AI pharma will be significantly accelerated. However, the application and diffusion of any technology are challenging to achieve overnight, and it is the law of development that new things spiral and move in waves. AI and data-driven pharma models need to be explored and practiced more and more before they can truly demonstrate their value.