
Abstract
Statins are widely used and have been proven to be effective in the prevention of atherosclerotic vascular disease events, primarily by reducing plasma low-density lipoprotein cholesterol concentrations. Although statins are generally well tolerated and present an excellent safety profile, adverse effects from muscle toxicity and liver enzyme abnormalities may occur in some patients. Myopathy and rhabdomyolysis are rare with statin monotherapy at the approved dose ranges, but the risk increases with use of higher doses, interacting drugs and genetic predisposition. Asymptomatic increases in liver transaminases with statin treatment do not seem to be associated with an increased risk of liver disease. Therefore, statin treatment can be safely used in patients with mild to moderately abnormal liver tests that are potentially attributable to nonalcoholic fatty liver disease and can improve liver tests and reduce cardiovascular morbidity in this group of patients. The risks of other unfavorable effects such as the slightly increased risk of new-onset diabetes and potentially increased risk of haemorrhagic stroke are much smaller than the cardiovascular benefits with the use of statins.
Introduction
Statins, or 3-hydroxy-methylglutaryl coenzyme A reductase inhibitors, are a widely used group of drugs which are effective in reducing atherosclerotic cardiovascular disease (CVD) events, largely by reducing plasma low-density lipoprotein cholesterol (LDL-C) concentrations. Since the first statin, lovastatin, was introduced in 1987, the safety and the efficacy of this class of drugs have been studied extensively. Multiple large randomized controlled trials enrolling a wide variety of patients have demonstrated that statins significantly reduced CVD morbidity and mortality. Every 1 mmol/l reduction in LDL-C with statin therapy is associated with a proportional reduction of about 20% in major vascular events defined as coronary death, nonfatal myocardial infarction, coronary revascularization, or stroke [Baigent et al. 2005] and more intensive statin therapy with greater reductions in LDL-C appears to be associated with additional cardiovascular benefits [Baigent et al. 2010; Josan et al. 2008]. The recent meta-analyses have also dispelled any remaining doubts about increased risks of cancer or other nonvascular mortality with statin treatment, as there was no significant increase in any of these with the standard or more intensive statin regimens. The proven effects of statins on reducing CVD risk have led to the widespread and increasing use of statins and the use of more intensive regimens. Hence, the safety and tolerability of this group of drugs is of great importance.
Although statins are generally well tolerated and present a good safety profile, adverse effects such as muscle toxicity and effects on liver enzymes may occur in some patients. These adverse effects have been well documented in recent reviews, with muscle-related toxicity receiving the greatest attention [Armitage, 2007; Beltowski et al. 2009; Brown, 2008]. This review provides an update on the safety of statins incorporating some of the more recent findings such as the meta-analyses that have shown that intensive statin therapy is associated with a slightly increased risk of the development of diabetes [Sattar et al. 2010].
Muscle toxicity
Muscle toxicity including myopathy and rhabdomyolysis is the most significant and well documented adverse effect with statins. Muscle symptoms may vary from mild problems, for example, pain, tenderness or weakness with or without creatine kinase (CK) elevation, to the most severe condition of rhabdomyolysis [Armitage, 2007]. Rhabdomyolysis is a potentially life-threatening myopathy involving muscle breakdown and myoglobin release into the circulation causing a brown discoloration of urine and risk of acute renal failure [Armitage, 2007]. Rhabdomyolysis is rare, but milder degrees of myopathy are more common and myalgia referring to muscle pain with no rise in CK level to greater than 10 times the upper limit of normal (ULN) may occur in up to 5% of patients [Thompson et al. 2003]. Various definitions have been proposed for the different types of muscle damage and the terminology proposed by the American College of Cardiology/American Heart Association/National Heart, Lung and Blood Institute Clinical Advisory is shown in Table 1 [Pasternak et al. 2002].
Definitions for statin-related myopathy proposed by American College of Cardiology/American Heart Association/National Heart, Lung and Blood Institute Clinical Advisory [Pasternak et al. 2002].

US Food and Drug Administration proposed definition: CK > 50 times ULN and evidence of organ damage, such as renal compromise.
CK, creatine kinase; ULN, upper limit of normal.
All statins can cause muscle toxicity if high enough doses are used or drug interactions occur [Brewer, 2003] but the degree of risk within the therapeutic dose range may vary among statins. A previously approved statin drug, cerivastatin, was withdrawn from the market due to a greater incidence of fatal rhabdomyolysis than seen with other statins [Armitage, 2007; Bays, 2006]. The differences in safety risk are likely to be related to the statin dosage, the physicochemical properties and pharmacokinetics profiles of statins, the concurrent use of agents having a potential for drug interaction and genetic factors [Bays, 2006; Bellosta et al. 2004; Link et al. 2008; Peters et al. 2009]. However, the risk of myopathy does not appear to be significantly different among all the other statins currently available (except for simvastatin at the maximal dose of 80 mg) [Backes et al. 2009; Fung and Crook, 2011]. Moreover, some statins minimally metabolized by cytochrome P450 (CYP) enzymes, for example pitavastatin, are expected to have a lower risk of drug–drug interaction and related adverse drug reactions than other statins for which the disposition is dependent on CYP enzymes [Corsini and Ceska, 2011].
In randomized controlled trials of standard dose statin therapy (i.e. atorvastatin 10–20 mg, fluvastatin 40–80 mg, lovastatin 20–40 mg, pravastatin 40 mg, rosuvastatin 10 mg and simvastatin 20–40 mg), there was a very low risk of myopathy (<0.01%) [Armitage, 2007] with an incidence of myopathy similar to that of placebo [Abd and Jacobson, 2011; Gillett and Norrell, 2011]. However, the incidence of statin myopathy in observational studies is much higher than in clinical trials as patients at higher risk of statin-related adverse events (e.g. those with previous statin intolerance, those receiving interacting medications or those with other risk factors for rhabdomyolysis) are likely to be excluded from clinical trials [Abd and Jacobson, 2011]. It has been shown that myalgia can occur in up to 10% of patients prescribed statins in observational studies, but rhabdomyolysis continues to be rare [Joy and Hegele, 2009].
High doses
The risk of statin-induced muscle toxicity is more likely with higher doses and this appears to be more evident with the simvastatin dose of 80 mg [Armitage, 2007; Thompson et al. 2006]. In the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) trial, in which 12,064 patients with a history of myocardial infarction were treated with simvastatin 20 mg or 80 mg daily (mean follow up 6.7 years), the incidence of myopathy (defined as CK > 10 times ULN plus unexplained muscle symptoms) and rhabdomyolysis (defined as myopathy with CK > 40 times ULN plus evidence of end-organ damage) in patients on 80 mg/day was approximately 0.9% and 0.4%, respectively, compared with 0.02% and 0% for patients on 20 mg/day [Armitage et al. 2010]. The risk of myopathy and rhabdomyolysis with simvastatin 80 mg was highest during the first year of treatment. It was often the result of interactions with certain drugs and was frequently associated with a genetic predisposition toward simvastatin-related myopathy as discussed below [Armitage et al. 2010; Link et al. 2008].
Due to the increased risk of myopathy and rhabdomyolysis, the US Food and Drug Administration (FDA) has recently recommended limiting the use of the highest approved dose of simvastatin (80 mg) [FDA, 2011]. The advice regarding the dose of simvastatin with interacting drugs was also revised. The FDA recommended that healthcare professionals can maintain patients on treatment with simvastatin 80 mg only if they have been taking this dose for 12 months or more without evidence of muscle toxicity but not to start any new patients on this dose. For patients unable to achieve their LDL-C goal using the 40 mg dose of simvastatin, an alternative lipid-lowering treatment (instead of uptitration to the 80 mg dose) that provides greater LDL-C lowering should be given [FDA, 2011].
The safety of higher doses of atorvastatin has been evaluated in more than 11,000 patients and the rate of clinically significant myopathy was very low [Waters, 2005]. Randomized controlled trials and meta-analyses revealed that there was a lack of dose dependency of the muscle adverse events of atorvastatin (10 mg versus 80 mg) [Athyros et al. 2010b; LaRosa et al. 2005; Newman et al. 2006]. In a meta-analysis of 49 clinical studies involving more than 14,000 patients treated with atorvastatin (10 or 80 mg/day) or placebo, treatment-associated myalgia was observed in 1.4%, 1.5% and 0.7% of patients on atorvastatin 10 mg/day, atorvastatin 80 mg/day and placebo, respectively, and there were no cases of rhabdomyolysis in these studies [Newman et al. 2006]. However, an observational study in an unselected population of 7924 patients with hyperlipidemia receiving high-dose statin therapy in general practice in France reported that 14.9% of patients on atorvastatin 40–80 mg/day experienced muscular symptoms [Bruckert et al. 2005]. This was comparable to those seen with simvastatin 40–80 mg/day, pravastatin 40 mg/day or fluvastatin 80 mg/day (18.2, 10.9 and 5.2%, respectively) in the same study, but the study did not include cases with asymptomatic CK elevations [Bruckert et al. 2005]. The incidence of muscle-related adverse events was considered to increase approximately fourfold to fivefold when the dose of atorvastatin was increased from 40 to 80 mg/day [Athyros et al. 2010b; Davidson, 2002].
Attention was drawn to potential safety concerns regarding muscle and renal damage when rosuvastatin was first marketed [Wolfe, 2004], but evidence from both clinical trials and postmarketing surveillance in a broad range of patients has clearly demonstrated that rosuvastatin in doses up to 40 mg has no excess muscle toxicity compared with other statins [Armitage, 2007; Shepherd et al. 2007; Toth and Dayspring, 2011]. In placebo-controlled trials, myopathy possibly related to treatment was reported in up to 0.1% of patients taking rosuvastatin at doses of up to 40 mg and higher doses appeared to be associated with increased risk of myopathy [Armitage, 2007; Shepherd et al. 2007]. The incidence rates of myopathy and rhabdomyolysis with pravastatin and pitavastatin appear to be comparable to other statins, with higher doses associated with increased risk but the high doses of these statins have not been well assessed in large randomized trials [Alsheikh-Ali and Karas, 2007; da Silva, 2011; del Sol and Nanayakkara, 2008].
Drug interactions
Drugs which interfere with statin metabolism or transport leading to increased systemic exposures to the drug increase the risk of myopathy, as summarized in Table 2 [Bellosta et al. 2004; Bottorff, 2006; Chatzizisis et al. 2010; Frishman and Horn, 2008]. It has been estimated that approximately 60% of cases of statin-related rhabdomyolysis are related to drug interactions [Chatzizisis et al. 2010; Kashani et al. 2006]. Many of the statin–drug interactions involve the CYP3A4/5 drug metabolizing systems as simvastatin, lovastatin and atorvastatin are metabolized predominantly through this pathway [Bellosta et al. 2004; Bottorff, 2006; Chatzizisis et al. 2010; Frishman and Horn, 2008]. Simvastatin is likely to be more sensitive to CYP3A4/5 inhibition than atorvastatin because extensive first-pass metabolism results in a systemic bioavailability less than 5% for simvastatin compared with 12% for atorvastatin [Neuvonen et al. 2006]. Some interactions may also be modulated by the efflux transporter P-glycoprotein (P-gp, gene name ABCB1), which shares many common substrates and inhibitors with CYP3A enzymes [Holtzman et al. 2006]. Fluvastatin is metabolized primarily by CYP2C9, with CYP3A4 and CYP2C8 contributing to a lesser extent, whereas pravastatin, rosuvastatin, and pitavastatin undergo little metabolism via CYP pathways and are excreted mainly unchanged, although rosuvastatin undergoes slight CYP2C9 metabolism [Bellosta et al. 2004; Frishman and Horn, 2008].
Selected drugs that may increase the risk of statin-induced myopathy and rhabdomyolysis.
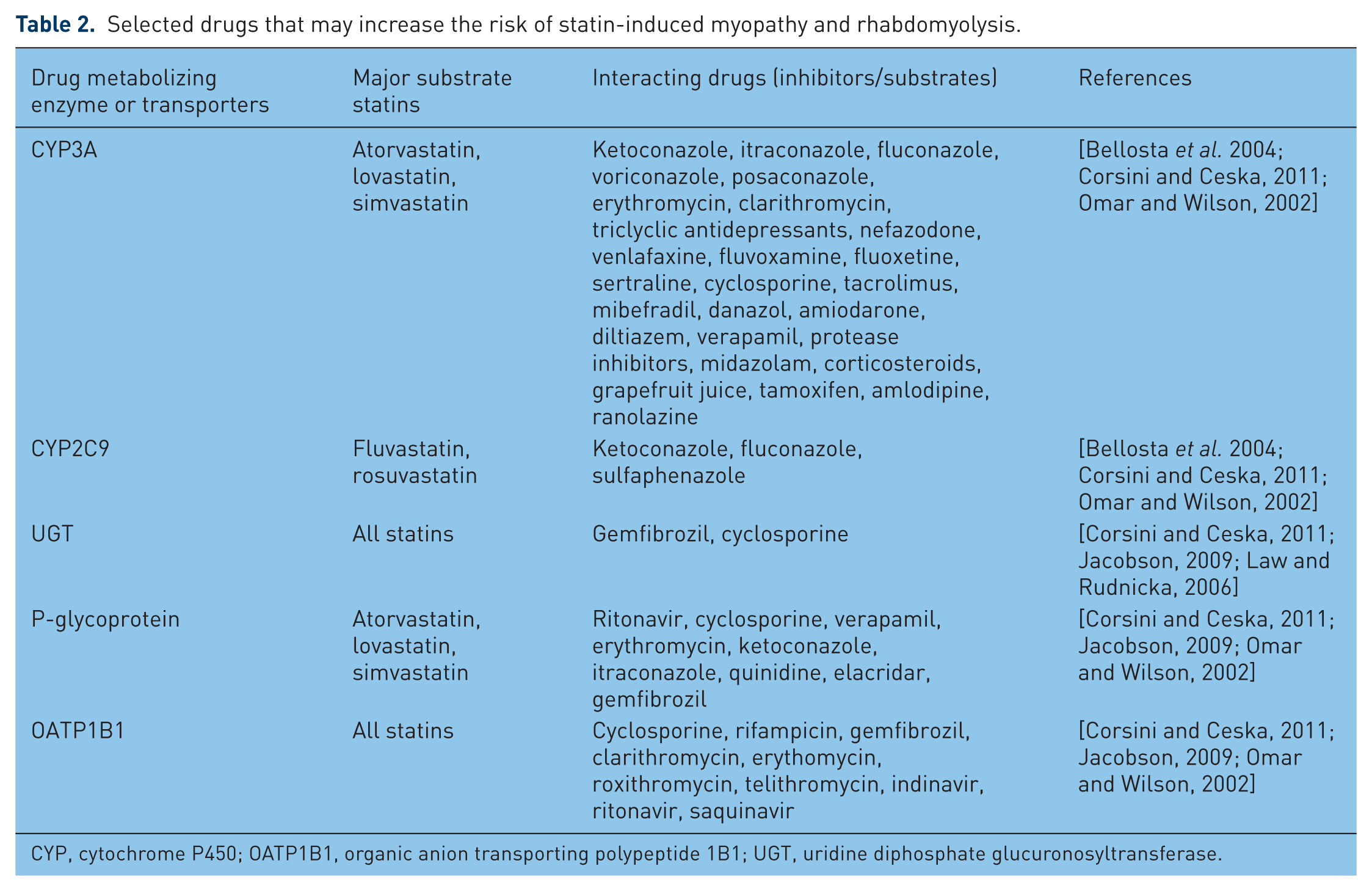
CYP, cytochrome P450; OATP1B1, organic anion transporting polypeptide 1B1; UGT, uridine diphosphate glucuronosyltransferase.
In addition to drug metabolizing enzymes, drugs interfering with the transporters responsible for hepatic uptake and excretion of statins may also affect the pharmacokinetics of statins and the risk of myopathy. The organic anion-transporting polypeptide (OATP) 1B1 (gene name SLCOB1) mediates the hepatic uptake of all statins to a variable extent and the efflux transporters P-gp and breast cancer resistance protein (BCRP, gene name ABCG2) are involved in the transport of most of the statins from the hepatocytes into the bile [Frishman and Horn, 2008; Ieiri et al. 2009]. Some drugs such as cyclosporine or clarithromycin will interact with statins through multiple pathways involving enzymes and transporters. Conversely, statins may cause interactions with other drugs resulting in toxicity. This is only a problem with drugs with a narrow therapeutic range, such as warfarin and digoxin. All of the statins can cause some increase in anticoagulation effect with warfarin, although the mechanisms are not clear except that fluvastatin can inhibit the metabolism of active S-warfarin through CYP2C9 [Neuvonen et al. 2006].
To avoid statin–drug interaction related myopathy, dose limits are set when prescribing statins with known potent inhibitors of the CYP enzymes or with some other interacting drugs. Due to the increased risk of myopathy with the high dose of simvastatin (80 mg), the FDA has recently recommended lower doses of simvastatin with certain CYP3A4 inhibitors, for example, the dose of simvastatin was limited to 10 mg daily in patients taking amiodarone, verapamil or diltiazem [FDA, 2011]. Furthermore, simvastatin is now contraindicated in patients taking gemfibrozil, cyclosporine and danazol, whereas previously the FDA recommended restricting the dose of simvastatin to up to 10 mg. Because of an increased risk of myopathy in Chinese patients taking simvastatin 40 mg administered with lipid-modifying doses (>1 g/day) of niacin-containing products observed in an ongoing, double-blind, randomized cardiovascular outcomes trial, caution is advised when treating Chinese patients with simvastatin doses exceeding 20 mg/day administered with niacin [FDA, 2010]. The disposition of pravastatin, rosuvastatin and pitavastatin does not depend on or is only marginally influenced by CYP enzymes and therefore these statins may have favorable drug interaction profiles [Corsini and Ceska, 2011; del Sol and Nanayakkara, 2008; Toth and Dayspring, 2011].
Pharmacogenetics of statin-induced myopathy
As statin-induced myopathy is a plasma- concentration-dependent adverse reaction, polymorphisms in CYP enzymes and drug transporters involved in the disposition of statins may affect the pharmacokinetics and toxicity of statins [Hu et al. 2011; Niemi, 2010; Peters et al. 2009; Romaine et al. 2009]. However, polymorphisms in CYP enzymes have been inconsistently associated with statin-induced myopathy and lipid responses [Fiegenbaum et al. 2005; Hu et al. 2009; Joy and Hegele, 2009; Peters et al. 2009; Wilke et al. 2005].
The uptake transporter OATP1B1 expressed in the basolateral membrane of hepatocytes regulates the influx of statins from the portal blood to the hepatocyte. The functional mutation in SLCO1B1 (521T>C) associated with reduced uptake activity of this transporter increases plasma levels of most statins [Niemi, 2010]. A genome-wide association study from the SEARCH participants taking the 80 mg dose of simvastatin and replicated in the Heart Protection Study with the 40 mg dose identified that the 521T>C in SLCO1B1 was significantly associated with simvastatin-induced myopathy [Link et al. 2008]. More than 60% of the myopathy cases with simvastatin 80 mg in the SEARCH trial could be attributed to the 521C variant. It has been suggested that this finding is likely to apply to other statins as SLCO1B1 polymorphisms affect the blood levels of most statins but the effect is greatest for simvastatin [Niemi, 2010; Pasanen et al. 2006; Romaine et al. 2009].
Most recently, a pharmacogenetic study examined whether common genetic polymorphisms that lead to reduced activity of CYP enzymes or transporters would be associated with mild statin-induced adverse effects defined as discontinuation for any side effect, myalgia, or CK more than three times ULN [Voora et al. 2009]. In this study, 509 patients were randomly assigned to 8 weeks of 10 mg/day atorvastatin, 20 mg/day simvastatin, or 10 mg/day pravastatin followed by 8 weeks of 80 mg/day atorvastatin, 80 mg/day simvastatin, and 40 mg/day pravastatin, respectively [Voora et al. 2009]. The study-defined adverse effects occurred in 99 patients (54 discontinuations, 49 myalgias, and 9 CK elevations) with a greater risk of the side effects with the high statin doses. The SLCO1B1 521T>C polymorphism was associated with statin-induced side effects in a gene dose-dependent manner, and the carriers of the 521C variant allele were at a twofold relative risk. Furthermore, this study also identified that the 521T>C polymorphism was associated with increased plasma levels of metabolites and increased risk of the adverse effects in patients assigned to simvastatin but not in those assigned to pravastatin (the plasma levels of atorvastatin metabolites were not assessed) [Voora et al. 2009]. Similar findings were also observed by Donnelly and colleagues in 4196 patients with type 2 diabetes receiving statins as part of routine clinical care [Donnelly et al. 2011]. This observational study demonstrated that the SLCO1B1 388A>G and 521T>C polymorphisms were associated with a lower (odds ratio 0.71, p = 0.026) and a higher (odds ratio 2.05, p = 0.043) incidence of statin intolerance, respectively, which was defined by biochemical abnormalities and also by discontinuation, switching, or reduction in dose of the prescribed statin drug, suggesting the role of SLCO1B1 polymorphisms in statin tolerability in the real world.
Other risk factors
There are multiple predisposing factors potentially increasing the risk of statin-induced myopathy, including advanced age, female sex, small body frame and frailty, alcoholism, complex medical problems (e.g. hypothyroidism or impaired liver/renal function), and history or family history of myopathy [Chatzizisis et al. 2010; Joy and Hegele, 2009; Thompson et al. 2003]. Furthermore, certain acutely acting factors such as the use of addictive drugs, serious viral or bacterial infections, major trauma, major surgery or the perioperative period, and intense physical activity could predispose patients to myopathy independently, and may trigger the development of severe myopathy in those receiving statins [Chatzizisis et al. 2010; Joy and Hegele, 2009].
It has been proposed that statin-induced coenzyme Q10 (CoQ10) deficiency might be involved in the pathogenesis of statin myopathy [Marcoff and Thompson, 2007]. CoQ10 plays an important role in mitochondrial energy production by participating in the electron transport chain, preventing oxidative stress, and regenerating active antioxidant vitamins C and E in mitochondria [Joy and Hegele, 2009; Marcoff and Thompson, 2007]. Polymorphisms of genes involved in the metabolism of CoQ10 have been inconsistently shown to be associated with statin-induced myopathy [Oh et al. 2007]. However, although statin treatment reduces circulating levels of CoQ10, largely because CoQ10 is carried in LDL particles, its effect on intramuscular levels of CoQ10 remains unclear. Furthermore, studies of the effects of oral CoQ10 supplementation which raise the plasma CoQ10 levels on myopathic symptoms are scarce and contradictory [Joy and Hegele, 2009; Marcoff and Thompson, 2007]. Owing to a lack of firm evidence, the routine use of CoQ10 was not recommended in patients receiving statin treatment, but CoQ10 supplementation can be tested in some patients who develop statin-related myalgia and who do not benefit from other approaches. Some patients may respond if only via a placebo effect, as shown in some preliminary trials [Young et al. 2007].
Vitamin D deficiency is associated with myositis and reduced muscle function [Erkal et al. 2006]. Serum 25(OH)D levels are related to physical performance in healthy people [Bunout et al. 2006]. Several recent case series and clinical studies demonstrated an association between vitamin D deficiency and statin-induced myalgia myopathy. Some studies have shown that statin-induced myositis myalgia in patients with low serum 25(OH)D levels was largely reversed (~90%) by vitamin D supplementation [Ahmed et al. 2009; Glueck et al. 2011a, 2011b; Lee et al. 2009]. These reports and studies suggested that vitamin D deficiency may contribute to the development of statin-induced muscle injury. Atorvastatin and rosuvastatin have been shown to increase serum 25(OH)D levels [Perez-Castrillon et al. 2007; Yavuz et al. 2009], which may contribute to their relatively lower incidence of myotoxicity than simvastatin, which does not influence vitamin D levels [Rejnmark et al. 2010]. However, future double-blind, placebo-controlled studies are required to verify observations on the relationship between serum vitamin D status and statin-induced myopathy.
Liver enzyme abnormalities
Elevation of liver enzymes (in particular, alanine and aspartate transaminases [ALT and AST]) is another well-recognized adverse effect of statins [Armitage, 2007; Gillett and Norrell, 2011]. Large randomized trials indicated that the dose-related elevations of ALT and AST levels observed in patients taking statins did not exceed those in patients taking placebo and little or no effect was seen on γ-glutamyltransferase, alkaline phosphatase, or bilirubin at low to moderate statin dosages [Argo et al. 2008; Armitage, 2007; Gillett and Norrell, 2011]. At the highest dose in the approved range, the incidence of the increases in ALT and AST is 20–30 per 1000 patients in comparison to 1–2 per 1000 patients with the lowest dose [Brown, 2008]. It has been estimated that the placebo-adjusted rise in transaminases attributable to statins was approximately 0.6 per 1000 patients [Brown, 2008; Law and Rudnicka, 2006].
The asymptomatic elevations in transaminases with statins generally occur in the first several months of treatment and are reversible. They can return to normal on stopping statin treatment, with dose reduction, or even spontaneously with continuation of the same statin dosage [McKenney et al. 2006]. It has been questioned whether the effect on transaminases indicates hepatotoxicity or just a hepatic reaction to a greater reduction in lipid levels as all lipid-lowering drugs may increase liver enzymes [Armitage, 2007]. Furthermore, emerging data have demonstrated that statins are generally well tolerated in patients with chronic liver disease with elevated liver enzyme levels, such as nonalcoholic fatty liver disease (NAFLD), primary biliary cirrhosis, and due to hepatitis C virus [Argo et al. 2008; Musso et al. 2011; Tandra and Vuppalanchi, 2009; Zamor and Russo, 2011].
Several short-term studies have evaluated statin treatments in patients with NAFLD and results have been promising, with some studies showing histological improvement in inflammation without changes in fibrosis [Argo et al. 2008]. Further evidence of the safety and efficacy of statins in patients with chronic liver disease was demonstrated in two recent clinical studies with pravastatin and atorvastatin, respectively [Athyros et al. 2010a; Lewis et al. 2007]. In a prospective, double-blind study of 326 patients with chronic liver disease, including 64% with NAFLD and 23% with hepatitis C, pravastatin 80 mg effectively reduced plasma LDL-C levels and fewer patients in the pravastatin group had elevations in ALT compared with the placebo group, although the difference was not statistically significant (7.5% versus 12.5%, p = 0.13) [Lewis et al. 2007]. The recent post hoc analysis of safety and efficacy of atorvastatin in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) study demonstrated that atorvastatin treatment significantly improved liver enzymes in patients with abnormal elevation in AST or ALT levels of up to three times the ULN at baseline during a 3-year follow up (−35%, −47% and −46% in ALT, AST, and γ-glutamyltransferase, respectively), whereas patients with abnormal liver enzymes who did not receive a statin showed a further rise in these levels during follow up [Athyros et al. 2010a]. Moreover, statin treatment reduced the risk of cardiovascular events by 68% in patients with abnormal liver tests and this benefit was greater than that seen in those with normal liver tests.
Risk of incident diabetes
The Justification for the Use of Statins in Primary Prevention: an intervention Trial Evaluating Rosuvastatin (JUPITER) study reported in 2008 showed an increased incidence of diabetes with rosuvastatin treatment and this reignited interest in the link between statin therapy and diabetes [Ridker et al. 2008]. A subsequent meta-analysis of 13 major placebo-controlled statin trials (including JUPITER) with 91,140 participants demonstrated a 9% increased risk of incident diabetes over 4 years (odds ratio 1.09; 95% confidence interval [CI] 1.02–1.17) in patients treated with statins compared with those receiving placebo [Sattar et al. 2010]. There was little heterogeneity among the studies included in the meta-analysis, suggesting that this risk appears to be a class effect [Sampson et al. 2011; Sattar et al. 2010]. The meta-regression analysis showed that the risk of development of diabetes with statins was highest in trials with older participants, but baseline body mass index and change in LDL-C concentrations were not found to be associated with the risk of new-onset diabetes [Sampson et al. 2011; Sattar et al. 2010]. The analysis suggested that treatment of 255 (95% CI 150–852) patients with statins for 4 years resulted in one extra case of diabetes [Sattar et al. 2010].
A more recent meta-analysis of five large (n > 1000) randomized controlled endpoint trials compared intensive-dose statin therapy with moderate-dose statin therapy in 32,752 participants without diabetes at baseline. The results showed that patients receiving intensive-dose statin therapy had increased risk of new-onset diabetes but a reduced risk of cardiovascular events compared with moderate-dose therapy over a weighted mean follow up of 4.9 years, with odds ratios of 1.12 (95% CI 1.04–1.44) and 0.84 (95% CI 0.75–0.94), respectively [Preiss et al. 2011]. Compared with moderate-dose statin therapy, the number needed to treat per year for intensive-dose statin therapy was 498 for the harm of new-onset diabetes while the number needed to treat per year was 155 for the benefit of reducing cardiovascular events.
Although statin therapy in general and intensive statin therapy in particular is associated with a slightly increased risk of developing diabetes, this risk was very small in absolute terms and relative to the reduction in coronary events [Preiss et al. 2011; Sampson et al. 2011; Sattar et al. 2010]. When comparing statins with some other cardiovascular drugs known to increase the risk of diabetes, such as β-blockers and thiazide diuretics, statins are much less likely to cause diabetes (odds ratio of incidence of diabetes for statins versus β-blockers and thiazide diuretics 1.09 [95% CI 1.02–1.17] versus 2.22 [95% CI 1.39–3.57] and 1.43 [95% CI 1.39–3.57], respectively) [Bhatia and Byrne, 2010; Elliott and Meyer, 2007]. Furthermore, the benefit of statins in preventing cardiovascular events in patients with diabetes has been well established [Kearney et al. 2008]. Thus, the cardiovascular benefits of statins are much greater than the small risk of incident diabetes in patients with moderate or high cardiovascular risk or with documented CVD [Preiss and Sattar, 2011; Sampson et al. 2011].
Haemorrhagic stroke
Previous studies suggested a close association between high serum cholesterol levels and stroke incidence [Athyros et al. 2010c; Tirschwell et al. 2004]. Statin therapy is associated with significant reductions in primary and recurrent ischemic stroke, probably via its lipid-lowering and pleiotropic effects [Athyros et al. 2010c; Bersano et al. 2008; Rodriguez-Yanez et al. 2008]. A meta-analysis of data from statin and nonstatin (diet or other lipid-lowering agents) trials demonstrated that statins, but not other interventions, significantly decreased the risk of total stoke (odds ratio 0.85, 95% CI 0.78–0.92, p < 0.001) and the reduction in total stroke risk was proportional to the reduction of total cholesterol and LDL-C levels [De Caterina et al. 2010].
However, a reverse relationship between cholesterol level and haemorrhagic strokes has been reported in different populations [Noda et al. 2009; Tirschwell et al. 2004; Zhao et al. 2008] and some cohort studies have found a small (19%) increase in the occurrence of haemorrhagic stroke on statin therapy [Athyros et al. 2010c; Law et al. 2003]. The recent Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial showed that there was an increase in the risk of haemorrhagic stroke during treatment with atorvastatin 80 mg/day compared with placebo for a period of 4.9 years (2.3% versus 1.4%, p = 0.002), although the relative risk of fatal and nonfatal stroke was reduced by 16% with atorvastatin treatment [Amarenco et al. 2006]. Post hoc analysis of this study showed that this increased risk was primarily observed in older men with a history of haemorrhagic stroke and there was no association between baseline or on-treatment LDL-C levels and the risk of haemorrhagic stroke [Goldstein et al. 2008]. Similarly, the early Heart Protection Study also suggested an increased risk of haemorrhagic stroke during simvastatin therapy among patients with previous cerebrovascular disease with no such increase evident in patients free from cerebrovascular disease [Collins et al. 2004]. However, a recent large population-based observational study in older patients surviving from acute ischemic stroke showed that exposure to statins was not associated with subsequent intracranial hemorrhage [Hackam et al. 2012]. This result was supported by an extensive meta-analysis of 11 randomized or observational studies (including SPARCL) exclusively enrolling patients with cerebrovascular disease showing that there was no evidence that statins selectively increased the risk of intracerebral hemorrhage [Hackam et al. 2011]. The other major statin trials in patients free from cerebrovascular disease did not show an increased risk of haemorrhagic stoke with cholesterol lowering [Amarenco and Labreuche, 2009; Everett et al. 2010]. The meta-analysis of the efficacy and safety of studies using more intensive lowering of LDL-C found a nonsignificant increased risk of haemorrhagic stroke with more intensive LDL-C lowering treatment compared with less intensive therapy, which was clearly offset by the reduction in ischemic strokes [Baigent et al. 2010; Josan et al. 2008]. Overall, it would appear prudent to be cautious in assessing the risks and benefits before using statins in patients with previous haemorrhagic stroke and to ensure that blood pressure is well controlled before giving very intensive statin therapy to avoid any increased risk of haemorrhagic stroke
Summary
Statins are a widely used and extensively studied group of drugs, which are well tolerated with good safety profiles. The recognized adverse effects of myopathy and rhabdomyolysis are rare at the approved dose ranges and the risk can be kept to a minimum by attention to the potential risk factors. Asymptomatic increases in liver transaminases with statin treatment does not seem to be harmful and statins can be safely used in most patients with elevated transaminase levels from NAFLD or stable hepatitis B and C viral infections. The risks of other unfavorable effects, for example, the slightly increased risk of new-onset diabetes and the potentially increased risk of haemorrhagic stroke, are small compared with the greater cardiovascular benefits with the use of statins.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Professor Brian Tomlinson has received research funding to perform clinical studies from AstraZeneca, Bayer, Boehringer Ingelheim, Daiichi Sankyo, Kowa, Merck Serono, Merck Sharp and Dohme, Novartis, Otsuka, Pfizer, Roche, Sanofi-Aventis and Servier. He has acted as a consultant or speaker on occasions for AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Kowa, Merck Serono, Merck Sharp and Dohme, Novartis, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, and Servier. The other authors have no known conflict of interest.