
Abstract
Although 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors are well tolerated, a small subset of patients may develop autoimmune myopathy, classified as immune-mediated necrotizing myopathy. Statin-induced immune-mediated necrotizing myopathy can present as proximal muscle weakness and in some cases as dysphagia and respiratory distress. We present two cases of patients taking statins who developed dysphagia and muscle weakness found to have statin-induced immune-mediated necrotizing myopathy. Both patients were treated with immunosuppressive therapy: one did well clinically, while the other had an aggressive form of statin-induced immune-mediated necrotizing myopathy and succumbed to the disease. Although statin-induced immune-mediated necrotizing myopathy is rare, early treatment to induce remission of this disabling condition should be initiated to prevent morbidity and mortality.
Introduction
Statins, 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors, are used ubiquitously to prevent morbidity and mortality from cardiovascular and cerebrovascular disease. While statins are largely well tolerated, a small subset of patients may develop autoimmune myopathy, characterized by proximal muscle weakness, elevated creatine kinase, and in some cases, dysphagia and respiratory distress. 1 Diagnosis of statin-induced immune-mediated necrotizing myopathy (SINAM) is made with positive autoantibodies to the target enzyme HMG-CoA reductase (HMGCR) and may need advanced imaging with biopsy. 2 While the classic symptoms are proximal muscle weakness, both our patients had dysphagia as the hallmark finding. It is pertinent to note that symptoms of SINAM vary from classic statin-induced myopathy and often times do not improve and might even worse after statin withdrawal. 3 We present two cases that highlight the presentation, diagnosis, and treatment of SINAM.
Case series
Case 1
A 60-year-old Hispanic man with a past medical history of hyperlipidemia and diabetes mellitus presented with elevated liver enzymes incidentally found during an outpatient primary care visit. He reported a gradual onset of muscle weakness in his upper arms and thighs that began 3 months prior to presentation. He had difficulty keeping his arms above his head, brushing his hair and teeth, and rising from a sitting position. He also noted progressive dysphagia, dyspnea on exertion, dry mouth and eyes, and 11 kg weight loss. He denied rash, joint swelling, and the use of new medications or herbal dietary supplements (HDS). He had been taking atorvastatin 20 mg for many years. His physical exam was notable for 4/5 bilateral upper and lower extremity muscle strength.
Laboratory values were significant for alkaline phosphatase (ALP) 58 (normal 25–100 U/L), aspartate transaminase (AST) 238 (normal 10–40 U/L), alanine transaminase (ALT) 407 (normal 9–46 U/L), and creatinine kinase (CK) 7228 (normal 38–174 U/L). Bilirubin, C-reactive protein (CRP), and erythrocyte sedimentation rate (ESR) were normal (Table 1). Further workup revealed negative hepatitis A, B, and C, antinuclear antibody (ANA), smooth muscle antibody, liver–kidney microsomal antibody, total IgG, ferritin, iron, total iron binding capacity (TIBC), anti-mitochondrial antibody, and alpha-1 antitrypsin. Antibodies to HMGCR were positive at 257 U/mL (normal < 20 U/mL). Abdominal ultrasound was unremarkable, and computerized tomography (CT) abdomen/pelvis showed mild gastroduodenal wall thickening and sigmoid colon thickening. An esophagogastroduodenoscopy (EGD) and colonoscopy were performed to evaluate for suspected malignancy, and both were unremarkable. Magnetic resonance imaging (MRI) pelvis and femur showed patchy and symmetric intramuscular edema of the anterior hip flexors, gluteal muscles, and adductor compartment muscles bilaterally (Figure 1). The patient underwent a confirmatory muscle biopsy which revealed myonecrosis consistent with immune-mediated necrotizing myopathy. He was treated with one infusion course of intravenous immunoglobulin (IVIG) while admitted. He was discharged on a prednisone taper, mycophenolate, and with monthly IVIG infusions. He has continued to show improvement upon regular rheumatology outpatient follow-up.
Initial laboratory values (CK, ALT, AST, aldolase, LDH, CRP, ESR, and HMGCr Ab level), type of statin, dosage, duration of use, and treatment.
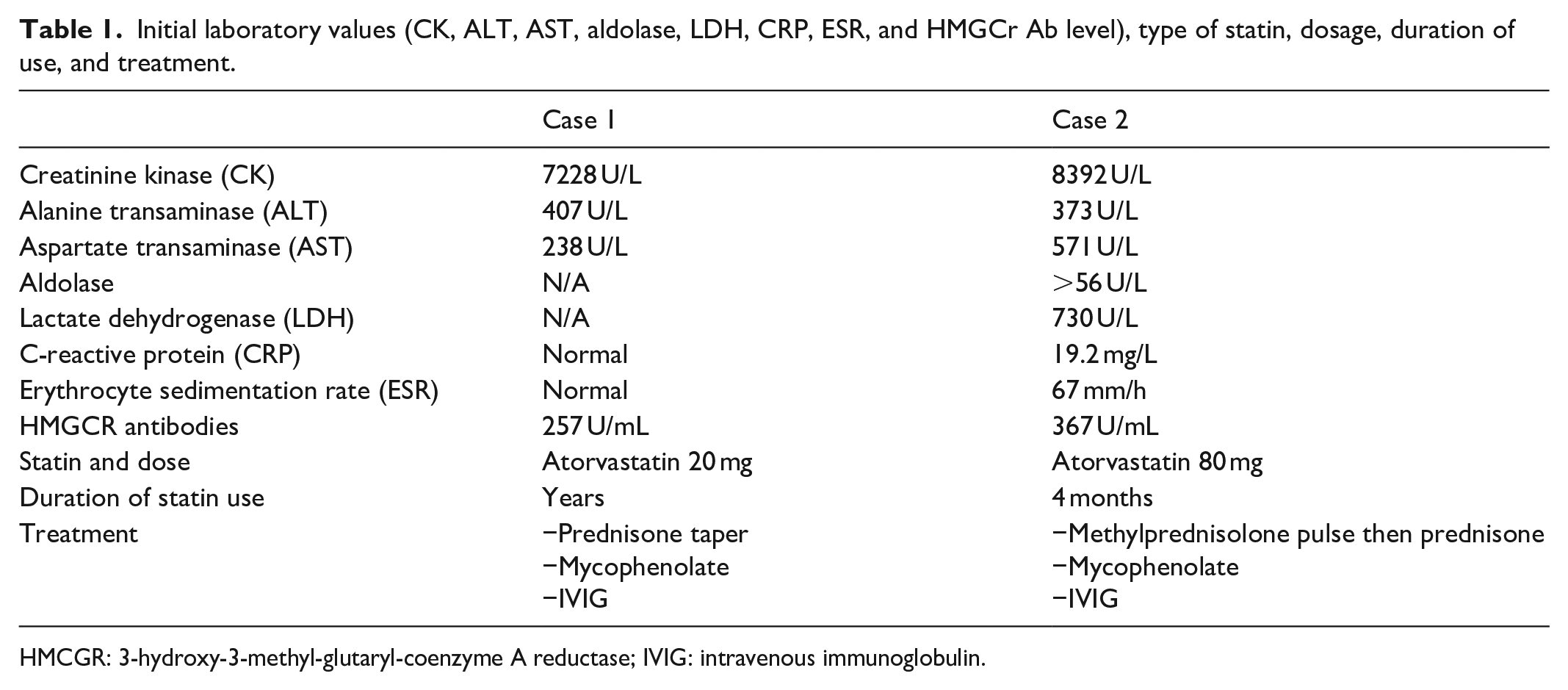
HMCGR: 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; IVIG: intravenous immunoglobulin.
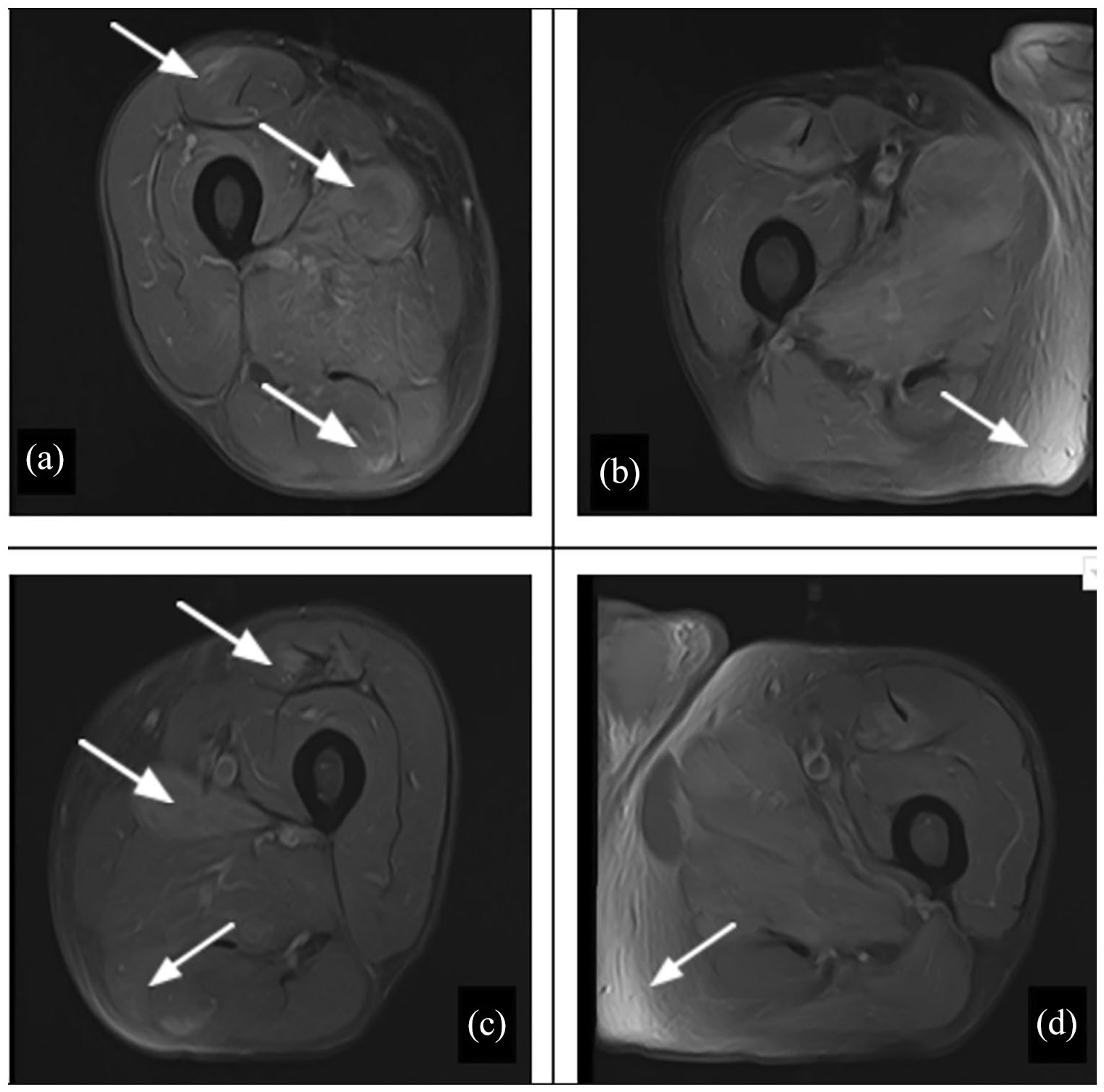
MRI of the right (a and b) and left (c and d) hip showing bilateral tissue inflammation and edema (arrows) in Case 1.
Case 2
A 71-year-old Hispanic man with a past medical history of hypertension, diabetes, and heart failure presented to the emergency department with worsening generalized weakness and dysphagia for the past month. He was experiencing difficulty raising his arms and legs and required maximal assistance to get out of bed. He also reported myalgia, severe dysphagia, and 13 kg weight loss. He denied recent travel and illicit drug use. He began taking atorvastatin 80 mg 4 months prior to admission. His physical exam revealed 3/5 muscle and neck flexor weakness. No rashes were appreciated, and a bedside swallow exam showed an inability to tolerate even ice chips.
Laboratory workup showed elevated ALP 79, AST 571, ALT 373, CK 8392, aldolase > 56 (normal ⩽ 8.1 U/L), lactate dehydrogenase (LDH) 730 U/L (105–333 U/L), CRP 19.2 (normal < 0.9 mg/dL), and ESR 67 (normal < 35.5 mm/h). A right upper-quadrant ultrasound showed diffuse echogenicity of the liver. Video fluoroscopy revealed silent aspiration with liquids and lack of cough reflex. Further serologic testing showed negative ANA and myositis panel including signal recognition protein (SRP), mildly elevated rheumatoid factor 25 IU/mL and positive HMGCR antibodies at 367 U/mL.
Given his striking weakness, dysphagia, and transaminitis, atorvastatin was held on admission due to concern for statin-induced myopathy. Rheumatology was consulted and recommended starting methylprednisolone even prior to return of serological workup, given strong clinical suspicion for SINAM. After confirmation with elevated HMGCR, IVIG was added. The patient was also started on prednisone and steroid-sparing immunosuppression with mycophenolate.
With treatment, the patient had gradual improvement of truncal stability and proximal strength. A downtrend of CK, ALT, and AST was also observed. Repeat video fluoroscopy showed mild improvement in swallowing, but there was persistent aspiration with liquid diet and a poor cough reflex. His hospital course was further complicated by acute hypoxic respiratory failure, pulmonary embolism, gastrointestinal bleed, and septic shock from aspiration pneumonia. Given his multiple complications and guarded prognosis, the patient’s family requested that the patient be discharged to hospice. He passed away shortly thereafter.
Discussion
The inflammatory myopathies are a heterogeneous family of diseases characterized by muscle weakness, elevated serum muscle enzyme levels, specific autoantibodies, and muscle biopsies confirming inflammation. In the last decade, muscle biopsies from patients suspected to have inflammatory myopathy have revealed myofiber necrosis and minimal to no lymphocytic infiltrates. This new pattern of myositis has been identified as immune-mediated necrotizing myopathy (IMNM). 1 IMNM is a rare type of autoimmune myopathy characterized by severe proximal weakness and infrequent extra-muscular involvement. Two different autoantibodies have been described in association with IMNM: SRP and HMGCR. Anti-HMGCR antibodies were discovered in 2010 and have since been strongly associated with SINAM. 2
SINAM incidence remains low with an estimated 2 to 3 cases in 100,000 patients taking statins per year. 2 Those with anti-SRP myopathy tend to be younger while those with anti-HMGCR myopathy tend to be older with a median age of 69.5. 4 There does not appear to be a clear ethnic predominance, but interestingly, both of our patients were Hispanic and had similar comorbidities. Moreover, the duration of statin use is not significant as myopathy can occur immediately after statin initiation or can occur years after. 5 Our patients were on statin therapy for many years and 4 months in each respective case. The exact pathogenesis of SINAM remains unclear. One of the theories is that statins may induce overexpression of HMGCR causing autoimmunity in genetically susceptible individuals. Despite statin discontinuation, the high HMGCR levels in regenerating muscle cells continue to drive autoimmunity once the response is activated. 2 The specific mechanism of muscle damage in SINAM is not fully understood, but a possibility may be that anti-HMGCR antibodies are pathogenic or may cross-react with different, unidentified antigens. 2
Statin-associated muscle symptoms can range in severity, from myalgia or myopathy to myositis and myonecrosis. 6 Dysphagia can occur as the initial or sole symptom. Patients with SINAM are more likely to have dysphagia than statin-naïve IMNM. 7 It is thought to be caused by inflammatory involvement of pharyngeal skeletal muscles which can lead to reduced pharyngeal contractility, cricopharyngeal dysfunction, and hypomotility of the esophagus. 8 The data on dysphagia are heterogenous but a meta-analysis by Labeit et al. 8 revealed a prevalence of 36%. Although muscular involvement beyond the limbs is uncommon, both of our patients also had dysphagia in addition to the usual proximal muscle weakness. Elevated laboratory values such as CK, AST, and ALT can aid in diagnosis, but testing for antibodies is crucial to establish the diagnosis. The strength of HMGCR as a screening and confirmatory tool for SINAM has been shown to be quite high. Shovman et al. 9 demonstrated high sensitivity and specificity for the assay, showing 92.3% and 100%, respectively. The diagnostic criteria most commonly used is European Neuromuscular Center (ENMC) criteria, which does not require a muscle biopsy to diagnose IMNM as serological studies alone can establish the diagnosis. 10 Only our patient in Case 1 received a biopsy. The clinical suspicion was high in Case 2 and confirmed with a positive HMGCR antibody level allowing us to defer a muscle biopsy. Furthermore, excluding occult malignancy in elderly patients with rapid onset and progressive myositis is crucial.
Due to paucity of cases, there are no clinical trials to guide therapeutic decisions. Therefore, treatment recommendations are based on case series and observational studies. The 2017 ENMC recommends induction steroids followed by maintenance immunosuppressive therapy. If patients do not achieve an adequate response, IVIG may be added, aside from consideration of starting rituximab. Early immunosuppressive therapy is the most effective induction therapy. 11 The goal for treatment is to induce complete remission, defined as normal strength and normalization of CK. However, treatment response tends to be heterogeneous, with half the patients having persistent weakness 2 years after treatment. 1 There is significantly more experience with the use of rituximab in SRP-autoantibody myositis, but its use is growing among SINAM cases as well. 8 Both of our patients received high-dose steroids but also required further therapy, given inadequate response. The patient in Case 1 received IVIG, mycophenolate, and a prednisone taper, while the patient in Case 2 received mycophenolate, IVIG, and methylprednisolone, given the persistence and severity of their symptoms. Both patients had long, protracted courses, with one eventually succumbing to the disease.
Conclusion
Although statin therapy has revolutionized the management and outcomes of atherosclerotic disease and is generally well-tolerated, it can still cause detrimental adverse effects. Clinical suspicion should be high for SINAM when patients present with proximal muscle weakness, dysphagia, elevated transaminases, and creatinine kinase. This series highlights the importance of speedy recognition of this clinical entity as early diagnosis of SINAM can allow for prompt initiation of treatment to prevent debilitating complications and mortality.
Footnotes
Author contributions
H.C., J.L., R.A., and J.H. reviewed the literature, drafted the manuscript, revised it for important intellectual content, and were involved in the final approval of the version to be published. C.Y. and M.R. revised the article for important intellectual content and were involved in the final approval of the version to be published.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent were obtained from the patient for Case 1 and from a legally authorized representative(s) for case 2 for their anonymized information to be published in this article.