
Abstract
Background:
Loss-of-function alterations in Makorin Ring Finger Protein 3 (MKRN3) and Delta-like Non-Canonical Notch Ligand 1 (DLK1) genes are associated with familial central precocious puberty (CPP) and can result in reduced serum levels of these proteins.
Objectives:
This study aimed to evaluate the potential of serum MKRN3 and DLK1 levels for predicting genetic variants associated with CPP.
Design:
Retrospective study.
Methods:
This retrospective cross-sectional study included 26 girls with CPP: 11 receiving treatment (Group 1) and 15 not yet treated (Group 2). The control group consisted of 26 healthy girls. Serum MKRN3 and DLK1 levels were measured by ELISA, and MKRN3 and DLK1 genes were analyzed by Sanger sequencing.
Results:
A known pathogenic MKRN3 gene variant (c.482dupC/p.(Ala162Glyfs*15)) was found in one patient with a notably low MKRN3 level of 0.127 ng/mL, consistent with paternal inheritance. Median MKRN3 and DLK1 levels were 1.32 (0.7) and 0.62 (0.4) ng/mL in Group 1, and 1.2 (0.6) and 0.62 (0.2) ng/mL in Group 2 (excluding the patient with the variant). No significant differences were observed between Group 1 and Group 2 (p = 0.279; p = 0.338). Control group median serum levels were 1.177 (0.76) ng/mL for MKRN3 and 0.67 (0.44) ng/mL for DLK1, with no significant differences from the CPP cohort (p = 0.917; p = 0.756).
Conclusion:
MKRN3 and DLK1 levels do not significantly differ between treated, untreated CPP patients, and healthy controls. Identifying a known MKRN3 variant with low serum levels suggests serum measurements can help predict variants. Familial studies are recommended as these variants may be inherited even in isolated cases.
Introduction
Central precocious puberty (CPP), characterized by early maturation of the hypothalamic-pituitary-gonadal axis, is defined as the onset of puberty before the age of 8 in girls and before the age of 9 years in boys. Many factors, such as hormonal, genetic, environmental, ethnic, nutritional, and socio-economic factors, play a role in the timing of the onset of puberty.1,2
The incidence of CPP is 10- to 20-fold higher in girls and varies widely across geographical regions, ranging from 0.217 to 26.28 per 10,000 girls and 0.02 to 0.9 per 10,000 boys. Large-scale studies from Spain, Denmark, and Korea have consistently shown a substantial increase in the incidence of CPP over the past two decades.3–6 Familial CPP accounts for approximately 27.5% of idiopathic cases. 6 Loss-of-function variants in the imprinted genes Makorin Ring Finger Protein 3 (MKRN3) and Delta-like Non-Canonical Notch Ligand 1 (DLK1) represent the most frequent monogenic causes of familial CPP.1,7–9
In a study from Denmark among healthy girls, a negative association between circulating MKRN3 level and gonadotropin concentration was demonstrated, which is the major inhibitor of gonadotropin-releasing hormones (GnRH). 10 However, Dauber et al. 7 reported the first loss-of-function variation of DLK1 supported by undetectable serum DLK1 levels in affected individuals.
Building on these reports, markedly low or undetectable serum levels may strongly indicate the presence of pathogenic variants in the genes mentioned above, as suggested by previous studies. In our study, we aimed to further investigate this association by evaluating the predictive value of variant detection in two specific genes, MKRN3 and DLK1, with serum levels.
Methods
Patients
Between March 2021 and September 2022, 26 girls with central precocious puberty diagnosed before 8 years of age and evaluated at our pediatric endocrinology outpatient clinic were enrolled in the study after obtaining informed consent. Patients with peripheral precocious puberty, underlying central nervous system pathology, chronic systemic disease, or incomplete medical data were excluded. Due to the retrospective nature of the study, a formal sample size calculation or power analysis was not performed. All eligible patients presenting during the study period were included, and, therefore, the sample size was determined by case availability. Among the study population, 11 patients were already receiving GnRH analog therapy at the time of enrollment (Group 1), whereas the remaining 15 were included at their initial clinical presentation and had not yet undergone treatment (Group 2). During follow-up, treatment was initiated in 12 of these patients; however, in 3 cases, GnRH analog therapy could not be initiated because the parents declined treatment. The control group consisted of 26 healthy girls who visited the pediatric outpatient clinic for routine follow-up and had no health issues. The local medical ethics committee granted medical ethical approval (2022/879945). Height and body mass index (BMI) standard deviation score (SDS) values for all CPP groups and the control group were calculated using national reference data. 11 The onset of puberty was defined by breast development, 12 pubertal basal luteinizing hormone (LH) levels, and/or GnRH-stimulated responses (peak LH level greater than 5 IU/L in response to LH-RH). Bone age (BA) was evaluated using the Greulich and Pyle method. The predicted adult height (PAH) was calculated using the Bayley Pinneau method. 13 Pelvic ultrasound and hypothalamus-pituitary MRI were done to rule out the possible organic etiologies of precocious puberty. Pubertal findings on pelvic ultrasound were defined as: uterine volume >2.0 mL, longitudinal diameter >35 mm, presence of endometrial echo, and ovarian volume >2–3 mL. 14
Laboratory studies
MKRN3 and DLK1 levels were measured in baseline samples (taken before the initiation of leuprolide acetate treatment) from 15/26 girls; they were collected from 11 girls during the treatment. The serum levels of MKRN3 and DLK1 were measured using the commercially available MKRN3 and DLK1 ELISA kits from the Bioassay Technology Laboratory, Shanghai, China. All samples were analyzed in the same laboratory. The intra- and interassay variations of DLK1 were <8% and <10, respectively. The assay has a range of 0.02–6 ng/L and a sensitivity of 0.00947 ng/mL. The assay range for MKRN3 is 0.05–10 ng/mL with a sensitivity of 0.019 ng/mL.
Genetic studies
DNA isolations were performed using the MagPurix Kit 200; Zinexts Life Science Corp. (Medsantek, Istanbul) following the manufacturer’s instructions. Ensembl was utilized for human reference gene sequences to design the primers, and the primer specificity was verified through the UCSC in silico polymerase chain reaction program. 15 Amplification is performed in thermal cyclers (SimpliAmpTM, Applied Biosystems, Foster City, CA, USA; and DNA Engine T100TM; Bio-Rad Laboratories, Hercules, CA, USA). The coding regions of the MKRN3 (NM_005664.4) and DLK1 (NM_003836.7) genes were sequenced using the Sanger sequencing method in all girls who presented with CPP. The family segregation analysis was performed on a case with the identified variant.
Statistical analysis
Statistical analysis was performed using the Jamovi package program (version 2.3) (Computer Software), Sydney, Australia. The prevalence analysis stating frequency is indicated both as a number (n) and a percentage (%). Values are stated as median [interquartile range (IQR)]. The Mann–Whitney U test was used for between-group comparisons of quantitative data. The significance of a p-value is ⩽0.05.
This manuscript was prepared in accordance with the Standardized Reporting of Burden of Disease studies (STROBOD) statement reporting guideline for cross-sectional studies. 16 A completed checklist based on the reporting recommendations for the burden of disease study is included as Supplemental Material.
Results
The median age of pubertal onset was 7.0 (IQR; 0.75) years in Group 1, and 7.0 (IQR; 0.87) years in Group 2. Three patients (P13, P14, P22) presented with menarche (Table 1). The median age of menarche of the mothers was 11.25 (IQR; 1.37) years in Group 1 and 12 (IQR; 1.0) years in Group 2. There was no statistical difference in these parameters between the groups (p = 0.587 and p = 0.504, respectively). Additionally, a history of early puberty was observed in the family of four patients (P11, P13, P14, P15) in Group 1, and similarly, in the family of four patients in Group 2 (P16, P23, P24, P25). One of the patients in Group 1 (P1) had a family history of X-linked hypophosphatemic rickets. Her mother and brother had lower extremity deformities. One patient had autism and had been receiving melatonin treatment since the age of 3.5 years (P16). No organic pathology was detected in cranial imaging of any of the patients.
Anthropometric and laboratory findings of patients with puberty precocious at presentation.
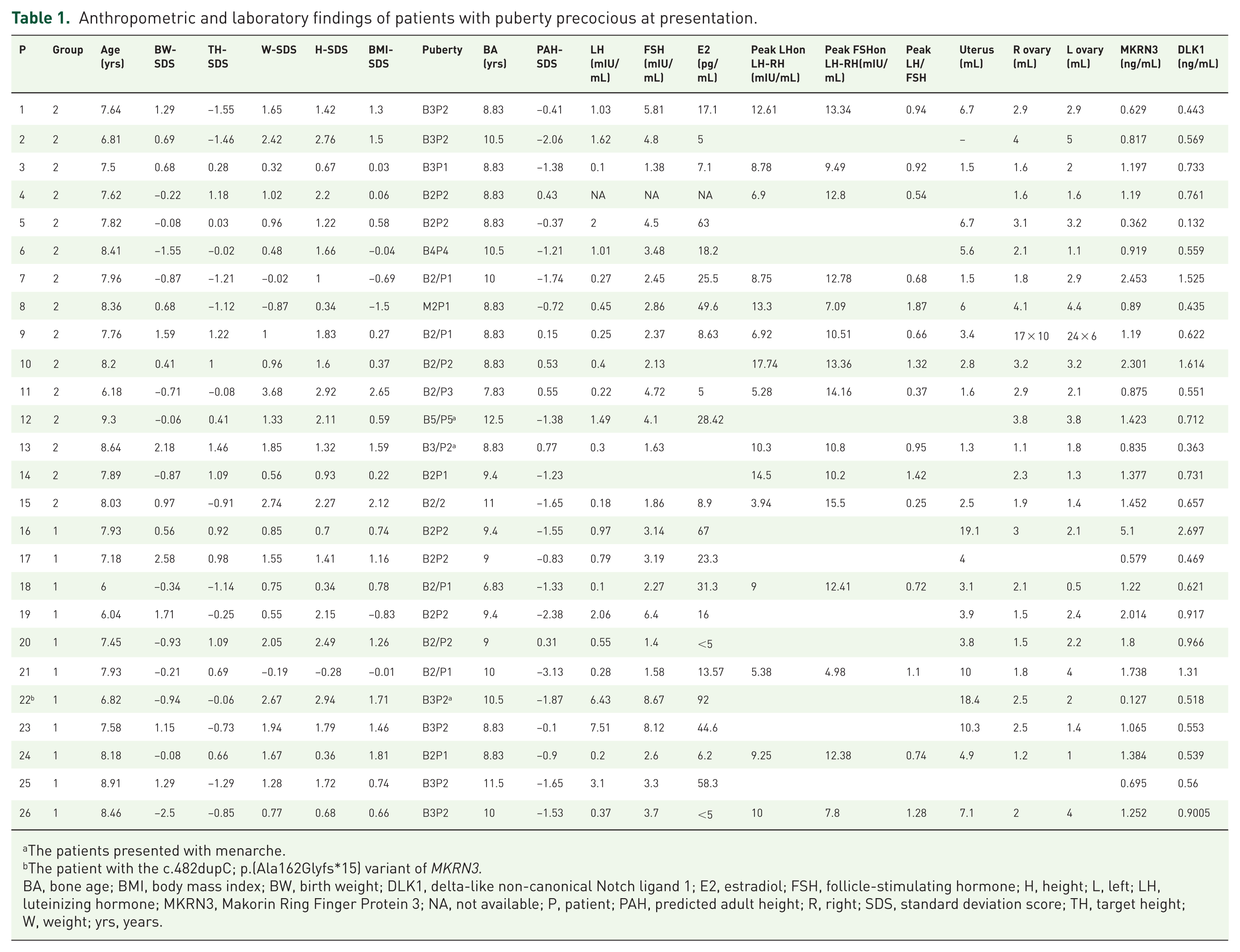
The patients presented with menarche.
The patient with the c.482dupC; p.(Ala162Glyfs*15) variant of MKRN3.
BA, bone age; BMI, body mass index; BW, birth weight; DLK1, delta-like non-canonical Notch ligand 1; E2, estradiol; FSH, follicle-stimulating hormone; H, height; L, left; LH, luteinizing hormone; MKRN3, Makorin Ring Finger Protein 3; NA, not available; P, patient; PAH, predicted adult height; R, right; SDS, standard deviation score; TH, target height; W, weight; yrs, years.
The details of the anthropometric and laboratory findings of patients at presentation and those who received treatment are shown in Tables 1 and 2. The comparison of the median age, height-SDS, BMI-SDS, target height-SDS, BA, and PAH-SDS of Group 1 and Group 2 was illustrated in Table 3, showing no statistically significant differences between the two groups. The comparison between the total CPP group and the control group revealed a significant difference in median age [7.8 (IQR; 0.7) 1.2 vs 7.2 (IQR; 1.2) ± 0.7 years; p = 0.04]. While the median height-SDS was significantly higher in the CPP group compared with the control group (p = 0.004), there was no significant difference in BMI-SDS between the two groups (p = 0.058; Table 3).
Anthropometric and clinical findings of patients with precocious puberty at the start of the treatment.

This patient’s mother has lower extremity deformities due to hypophosphatemic rickets.
BA, bone age; BMI, body mass index; H, height; NA, not available; P, patient; PAH, predicted adult height; SDS, standard deviation score; W, weight; yrs, years.
Comparison of anthropometric measurements and serum MKRN3 and DLK1 levels between groups.

Comparison of age, height, and BMI-SDS, serum MKRN3 and DLK1 levels in patients with CPP (Group1 and Group 2) and Control Group.
Comparison of age, BA, height, BMI, target height, and PAH-SDS, serum MKRN3, and DLK1 levels in Group1 and Group 2.
Except the patient with c.482dupC/p.(Ala162Glyfs*15) variant.
BA, bone age; BMI, body mass index; DLK1, delta-like non-canonical Notch ligand 1; IQR, interquartile range; MKRN3, Makorin Ring Finger Protein 3; PAH, predict adult height; SDS, standard deviation score.
In Group 2, a known heterozygous c.482dupC/p.(Ala162Glyfs*15) variant in MKRN3 was detected in P22. The segregation analysis of the MKRN3 variants in the family demonstrated adherence to the paternal inheritance model. Further analysis revealed that this variant was also present in the aunt and grandmother (Figure 1a). The serum MKRN3 level was 0.127 ng/mL, which was also significantly lower than that of other family members (Figure 1b).
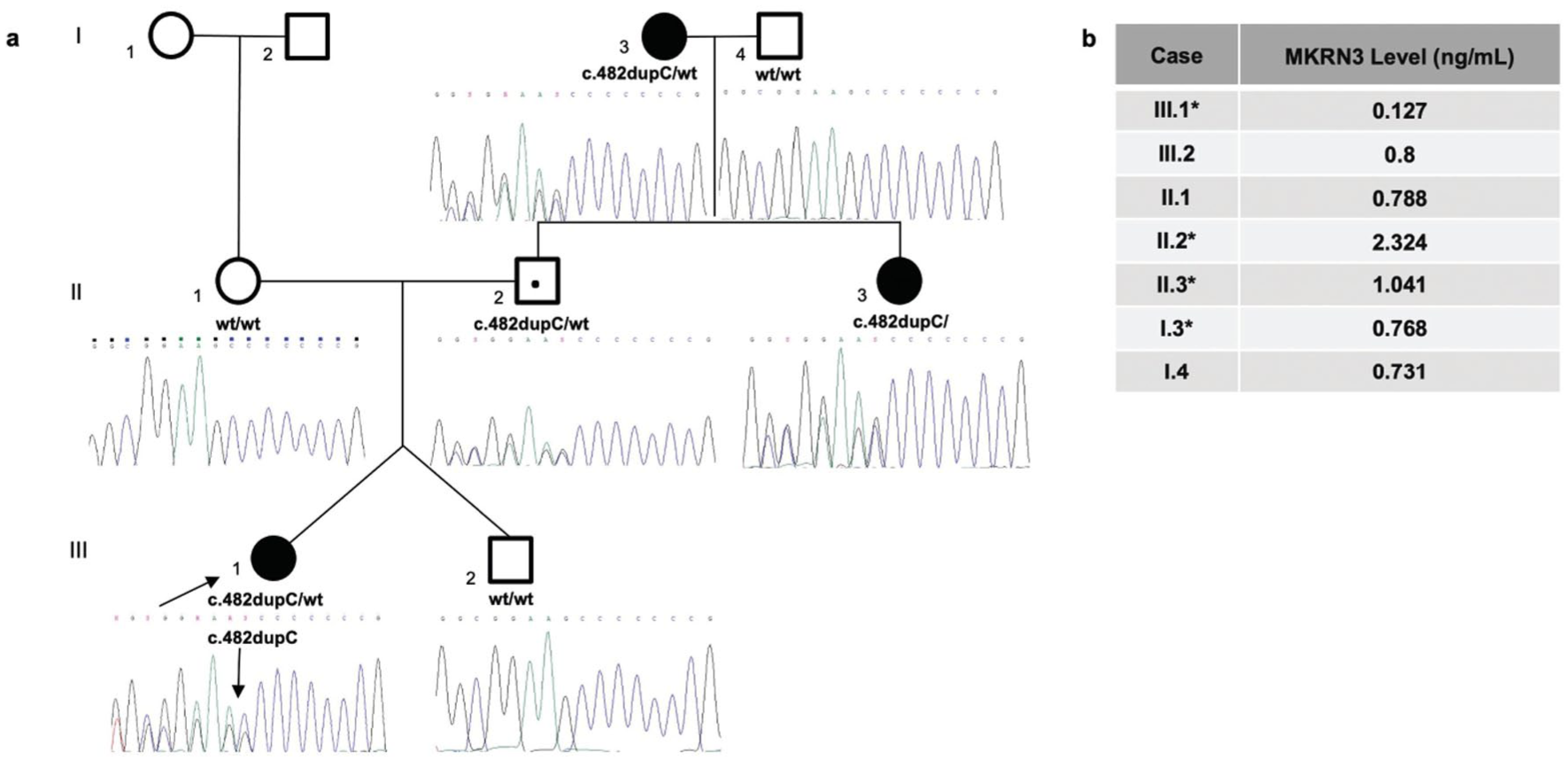
(a) Pedigree and sequencing data of patient (P22) with c.482dupC/p.(Ala162Glyfs*15), rs763195944 heterozygous variant. The arrow indicates probands, the open square indicates a male, and the open circle indicates a female. The black symbols represent affected individuals, and the black dots represent asymptomatic carriers according to the imprinted patterns of inheritance. (b) The serum MKRN3 level of the proband (III.1) was 0.127 ng/mL, which was significantly lower than that of other family members. The serum levels of other individuals who carried the variant were 2.324 (II.2), 1.041 (II.3), and 0.768 (I.3) ng/mL, respectively. The serum levels of other individuals who did not carry the variant were 0.8 (III.3), 0.788 (II.1), and 0.731 (I.4) ng/mL, respectively.
The median serum MKRN3 level was 1.32 (IQR; 0.7) and 1.2 (IQR; 0.6) in Group 1 and Group 2, respectively. The median serum DLK1 level was 0.62 (IQR; 0.4) and 0.6 (IQR; 0.2) in Group 1 and Group 2, respectively. There was no statistical difference in serum MKRN3 and DLK1 levels between groups (p = 0.279; p = 0.338). The serum data of the patient with the MKRN3 pathogenic allelic variant were excluded from these analyses. Regarding these parameters, there was no statistically significant difference between the total CPP group and the control group (p = 0.917; p = 0.756; Table 3). While the serum MKRN3 level was 0.127 ng/mL in the patient with the MKRN3 variant (c.482dupC; p.(Ala162Glyfs*15)), the median MKRN3 serum level was 1.197 (IQR; 0.57) ng/mL in the other girls with CPP.
Discussion
In this study, we evaluated serum levels of MKRN3 and DLK1 in girls with CPP, both before and during the treatment, and compared them to levels in healthy girls. We focused on predicting variants in the MKRN3 and DLK1 genes based on serum levels, investigating the correlation between serum measurements and these two genes. In addition, a known variant of MRKN3 was identified.8,9,17
Dauber et al. first reported the identification of a genomic defect in DLK1 associated with isolated familial CPP. In the same study, they demonstrated undetectable circulating levels of DLK1 in affected individuals, suggesting that the genomic deletion leads to a complete lack of DLK1 production. 3 Karaman et al. 18 reported novel nonsense and an intronic variant on DLK1 and investigated the effect of these variants on DLK1 production by serum levels. This study further supported the hypothesis that pathogenic variants can be predicted by observing low serum levels.
In a longitudinal study, a decrease in serum MKRN3 levels before puberty was reported in 38 healthy prepubertal girls monitored from the prepubertal period to puberty. 10 Grandon et al. also reported that MKRN3 levels were significantly lower in girls with CPP compared to prepubertal age-matched girls. In addition, in another longitudinal study conducted by the same team, MKRN3 levels decreased with GnRH analog treatment in 15 girls with CPP who did not have the MKRN3 variant during the treatment.19,20 However, in our recent study, we did not observe a statistical difference in MKRN3 levels between girls with CPP and healthy prepubertal girls. Although our study included girls in a similar age range as in previous studies,19,20 the possible explanations for the lack of differences in serum levels between girls with CPP and prepubertal girls, as well as on GnRHa treatment of the CPP girls, may include variations in the assay used or changes in the timing of studies after sample storage, as previously suggested. 20 One limitation of our study was the inability to longitudinally measure serum levels both before and during the treatment in patients, unlike the previous study. 20 Although it is known that MKRN3 levels decrease at the onset of puberty, the peripheral levels of MKRN3 may not be sufficient to evaluate the onset, progression, and treatment response of puberty, suggesting that epigenetic factors such as methylation differences may be effective. 21
Although the serum MKRN3 level was within the normal range according to the kit references, it was significantly lower in the patient carrying the c.482dupC/p.(Ala162Glyfs*15) variant in the MKRN3 gene compared to those patients with CPP and healthy girls, and also in family members. The affected amino acid in our case (Ala162Glyfs15) lies very close to the previously described pathogenic Pro160Cysfs14 variant, 21 which was associated with undetectable circulating MKRN3 levels. The clustering of disruptive variants within this region suggests that it may represent a critical functional domain of the MKRN3 protein. Although a different ELISA kit was used in the earlier study, 22 the markedly low MKRN3 level observed in our patient mirrors the biochemical pattern reported for Pro160Cysfs*14, reinforcing the likely functional importance of this domain. Additionally, despite markedly reduced or even undetectable serum MKRN3 levels being reported in patients with MKRN3 mutations, the researchers concluded that MKRN3 measurements cannot be recommended as a screening tool due to the large variability in serum concentrations. 22
The measurement of DLK1 is well established, with studies showing that gene loss-of-function correlates with low to undetectable serum levels, representing the cleaved form of the protein released into the bloodstream. 7 DLK1 also reflects pubertal stage, decreasing as puberty progresses in both girls and boys. 23 In contrast, serum MKRN3 measurement remains more controversial, as absolute levels do not consistently correlate with pubertal timing, and pathogenic variants can have unpredictable effects on circulating protein concentrations. On the other hand, the DLK1 gene sequencing analyzes five separate coding exons spanning 1149 bp (383 aa), which is technically challenging to analyze. The MKRN3 gene consists of a single coding exon spanning 1571 bp (507 aa), which, despite being a single exon, requires substantial effort to sequence. Considering our recent study and previous research on serum concentrations, as well as acknowledging the effort involved in genetic studies, conducting initial serum level examinations followed by genetic studies in patients with idiopathic CPP and low serum levels may represent a logical and cost-effective approach, allowing evaluation in larger cohorts and supporting the establishment of reliable cutoff values for future clinical use.
To date, no de novo MKRN3 variations have been described.1,24 In all patients with MKRN3 loss-of-function alterations, the segregation analysis was consistent with a paternal origin. Due to the imprinting pattern of MKRN3, clinical manifestations can be observed in girls carrying a pathogenic MKRN3 variation, even if their fathers are asymptomatic. 1 The familial segregation analysis of our case was supported by paternal inheritance. Our case’s familial segregation analysis supported paternal inheritance, as no early puberty history was present in other family members, and only the index case showed low serum MKRN3 levels. Although a variant was detected in only one patient, further genetic and/or epigenetic investigations may be necessary for patients with a family history of early puberty, even if no variants were identified.
Previous studies have reported that MKRN3 variants are relatively common, with a prevalence ranging from 9% to 46%.25,26 Karaman et al. 18 identified two DLK1 variants (11.1%) and one MKRN3 variant (5.5%). In this recent study, we detected only a single MKRN3 variant (3.8%). Overall, the frequency of pathogenic variants in MKRN3 and DLK1 across our two studies was 4.5%. In another study from Türkiye, a prevalence of pathogenic MKRN3 variants was reported in 2.9% of their cohort, while no DLK1 variants were identified in that study. 17
In our patient with the MKRN3 variant (P22), which can be described as deleterious and severe, we observed a higher basal LH level and advanced BA (Table 1). This observation is consistent with the findings of genotype–phenotype correlation studies, which suggest that deleterious and severe variants may result in a more pronounced clinical presentation. 24 Notably, compared to the patient reported by Kirkgoz et al. 17 with the same variant, our patient exhibited significantly more advanced BA and markedly higher basal LH levels.
Limitations and conclusion
One of the limitations of this study is the lack of an a priori sample size calculation or power analysis. Owing to the retrospective design, all eligible patients presenting during the study period were included, and the sample size was, therefore, determined by case availability; consequently, the number of patients was relatively small. Despite this limitation, assessing serum levels can be cost-effective and time-saving for identifying patients with CPP needing molecular analysis, particularly targeting those with low serum levels. Given the inheritance pattern, it is important to note that sporadic cases may also be familial. Performing segregation analysis is particularly important in cases where a variant has been identified.
Supplemental Material
sj-docx-1-tae-10.1177_20420188261424161 – Supplemental material for Use of serum MKRN3 and DLK1 levels in precocious puberty: association with an MKRN3 pathogenic variant
Supplemental material, sj-docx-1-tae-10.1177_20420188261424161 for Use of serum MKRN3 and DLK1 levels in precocious puberty: association with an MKRN3 pathogenic variant by Esin Karakilic-Ozturan, Volkan Karaman, Asuman Gedikbaşı, Şükran Poyrazoğlu, Feyza Darendeliler, Zehra Oya Uyguner and Firdevs Baş in Therapeutic Advances in Endocrinology and Metabolism
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.