
Abstract
Familial dysalbuminemic hyperthyroxinemia (FDH) is a form of euthyroid hyperthyroxinemia caused by ALB gene variants that is commonly misdiagnosed in clinical practice. When coexisting with papillary thyroid carcinoma (PTC), this condition may complicate postoperative management strategies. This study aims to enhance clinical recognition of the coexistence of FDH and PTC and explore evidence-based management approaches. We retrospectively analyzed the diagnostic and therapeutic course of a patient with concurrent FDH and PTC in our clinic and reviewed the current literature. The proband exhibited unsuppressed thyrotropin (TSH) by levothyroxine (L-T4) monotherapy after PTC surgery, with abnormal, persistently elevated total thyroxine (T4) and free T4 levels. Further review of the medical records revealed a similar pattern of thyroid function abnormalities preoperatively. Genetic testing confirmed FDH caused by an ALB gene c.725G>A mutation. Sanger verification confirmed that both the proband’s mother and daughter carried the same variant. The proband ultimately achieved adequate TSH suppression with the combination therapy of L-T4 and desiccated thyroid extract (DTE). This case highlights the importance of considering FDH when postoperative T4 and TSH levels show discordance in PTC patients. Increased albumin-T4 binding affinity in these patients may prevent adequate TSH suppression with L-T4 monotherapy, while DTE directly delivers triiodothyronine, without requiring peripheral conversion. Therefore, combination therapy with L-T4 and DTE may achieve effective TSH suppression in these patients.
Keywords
Introduction
Familial dysalbuminemic hyperthyroxinemia (FDH), first described in 1979, is an autosomal dominant disorder caused by mutations in the ALB gene that confer an increased binding affinity of albumin for thyroid hormones, thereby causing persistently elevated serum concentrations of thyroxine (T4) or triiodothyronine (T3). 1 Here, we report the first male case of FDH coexisting with papillary thyroid carcinoma (PTC) and review the relevant literature.
Case presentation
A 40-year-old male (the proband, II-2) was referred to our clinic in November 2022 due to persistent T4 elevation 3 months after thyroidectomy. A thyroid nodule was initially identified during a routine physical examination in June 2022. Subsequent ultrasonography at a local hospital revealed a 0.51 × 0.55 cm nodule (ACR-TIRADS 4) in the left thyroid lobe and a left level III cervical lymph node (1.0 × 0.4 cm) exhibiting punctate calcification. However, the subsequent contrast-enhanced CT scan of the neck revealed no evidence of lymph node metastasis. Fine-needle aspiration cytology of the thyroid nodule suggested PTC. On August 8, 2022, a left thyroid lobectomy with isthmusectomy, left radical neck dissection, and left recurrent laryngeal nerve exploration were performed. Histopathology confirmed classic PTC (maximal diameter 0.5 cm) in the left thyroid lobe with focal peri-thyroidal invasion but no definitive lymphovascular or perineural invasion. The surrounding thyroid tissue showed nodular goiter. Molecular testing identified a BRAFV600E mutation. No metastasis was found in the left central lymph nodes (0/3). The pathological stage was classified as pT1a(m)N0cM0 (AJCC 8th edition, Stage I), with intermediate recurrence risk according to the ATA guidelines. 2
Postoperatively, levothyroxine (L-T4) monotherapy was initiated at 75 μg daily. Thyroid function tests obtained on September 9, 2022 (postoperative day 32), revealed elevated total T4 (TT4; 233.33 nmol/L, normal range (NR) 78.38–157.40) and free T4 (FT4; 31.07 pmol/L, NR 7.67–16.25), with normal total T3 (TT3; 1.91 nmol/L, NR 1.02–2.48), free T3 (FT3; 5.08 pmol/L, NR 3.85–6.01), thyrotropin (TSH; 2.958 μIU/mL, NR 0.560–5.910), and unmeasurable thyroglobulin (Tg; <1 ng/mL, NR < 35). Thyroglobulin antibodies and thyroid peroxidase antibodies were both within the normal limits. Despite a gradual L-T4 dose reduction, persistently elevated FT4 and TT4 concentrations were observed (Table 1). A review of preoperative thyroid function records disclosed similar biochemical abnormalities, for which methimazole therapy (20 mg/day) had been administered at a local institution.
The thyroid function evolution and therapeutic regimen of the proband (II-2).

R: Roche; B: Beckman.
DTE, desiccated thyroid extract; FT3, free T3; FT4, free T4; L-T4, levothyroxine; T3, triiodothyronine; T4, thyroxine; Tg, thyroglobulin; TSH, thyrotropin; TT3, total T3; TT4, total T4.
Physical examination revealed unremarkable findings, with stable vital signs and no abnormalities detected on cardiac, pulmonary, or abdominal assessment. Pituitary evaluation revealed normal adrenal function: morning adrenocorticotropic hormone was 43.6 pg/mL (NR 7.2–63.3), serum cortisol 241.8 nmol/L (NR 170–440), 24-h urinary free cortisol 498 nmol/24 h (NR 108–961), and sex hormone-binding globulin 30.0 nmol/L (NR 18.3–54.1). Contrast-enhanced pituitary MRI demonstrated no structural abnormalities, excluding a TSH-secreting adenoma. Given the familial occurrence of similar thyroid dysfunction in the proband’s mother (I-2) and daughter (III-1; Table 2), whole-exome sequencing was performed on peripheral blood from the proband, identifying a heterozygous ALB gene mutation (c.725G>A, p.Arg218His) in exon 7. This variant corresponds to residue R218H in the mature protein following signal peptide cleavage and represents the most prevalent mutation associated with FDH. Sanger sequencing confirmed the variant in both affected relatives (Figures 1 and 2). No pathogenic variants were detected in THRA (Thyroid Hormone Receptor Alpha) and THRB (Thyroid Hormone Receptor Beta), effectively ruling out resistance to thyroid hormone (RTH) syndrome. In addition, no mutations were identified in the TTR (Transthyretin) or SERPINA7 (Serine Protease Inhibitor, A7) genes, thereby excluding genetic defects in other thyroid hormone transport proteins.
The thyroid function of the proband (II-2)’s family.
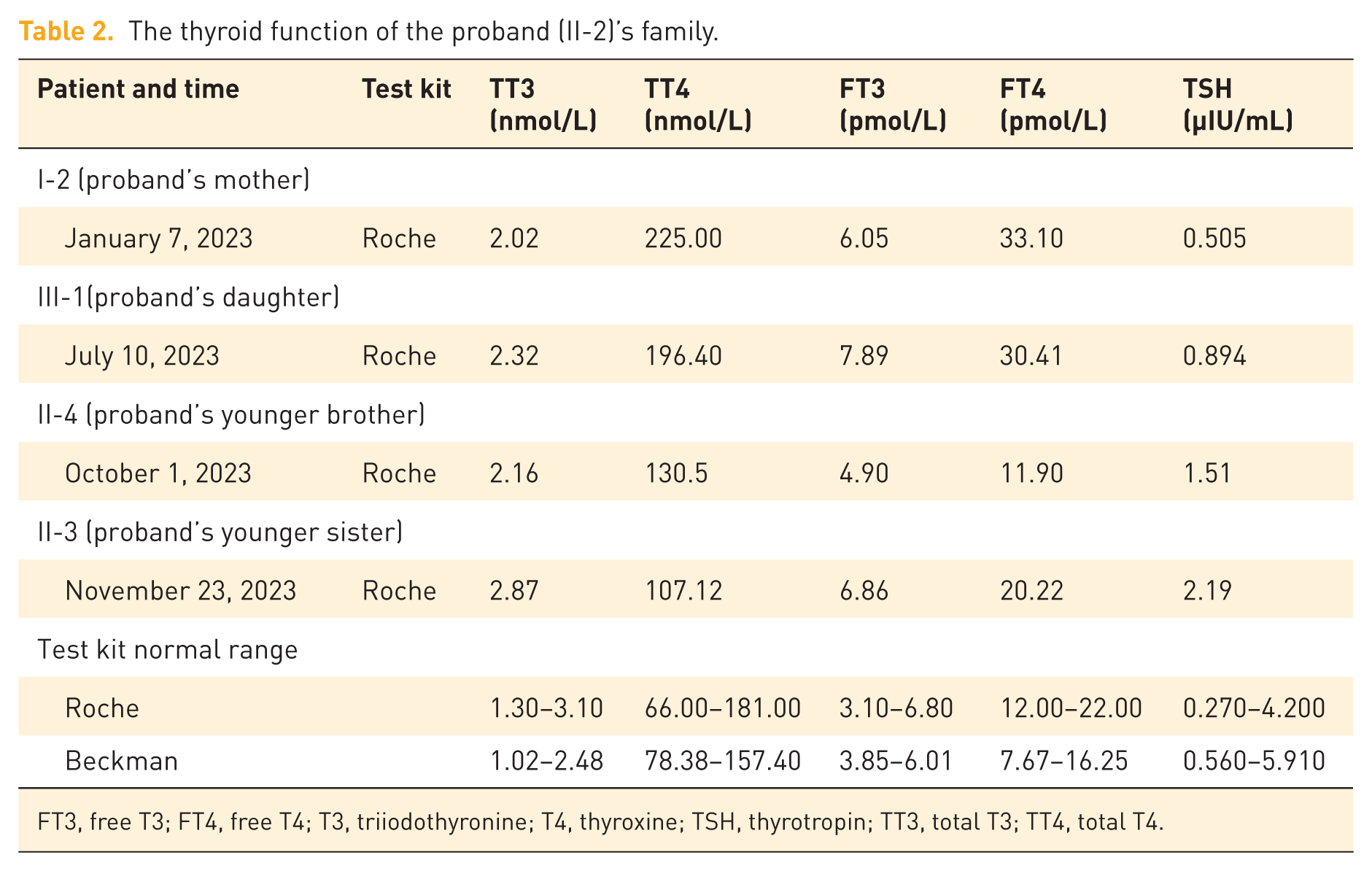
FT3, free T3; FT4, free T4; T3, triiodothyronine; T4, thyroxine; TSH, thyrotropin; TT3, total T3; TT4, total T4.

The ALB gene sequencing results for II-2 (a), I-2 (b), I-1 (c), III-1 (d), and II-3 (e). The c.725G>A (p.Arg218His) mutation (arrow) was identified in II-2 (a), I-2 (b), and III-1 (d), while this mutation was not detected in I-1 (c) and II-3 (e).

Family chart of ALB R218H/WT. Solid and open squares, respectively, represent male subjects ALB R218H/WT and ALB WT. Solid and open circles, respectively, represent female subjects ALB R218H/WT and ALB WT. Gray squares and circles represent male and female subjects who were not genetically tested but were euthyroid. Squares and circles containing question marks represent male and female subjects who underwent neither genetic testing nor thyroid function tests. The arrow indicates the proband (II-2).
Following confirmation of the genetic diagnosis, desiccated thyroid extract (DTE) was incorporated into the therapeutic regimen on January 5, 2023. A final stable regimen was established at L-T4 50 μg/day combined with DTE 40 mg/day. Subsequent thyroid function tests obtained on August 3, 2023, demonstrated a TSH level of 0.470 mIU/L, achieving the target TSH range for intermediate-risk PTC during initial postoperative management.
Discussion
In clinical practice, when elevated serum TT4 and FT4 concentrations coexist with normal TSH levels, clinicians should consider disorders such as syndrome of resistance to RTH, pituitary adenoma, FDH, or hereditary thyroxine-binding globulin abnormalities. We ruled out a pituitary adenoma by checking adrenal function and performing a contrast-enhanced pituitary MRI. The patient denied any thyroid-related symptoms and was taking no medications known to affect thyroid function tests. In addition, the family history, combined with the identification of a pathogenic variant in the ALB gene, helps exclude other possible conditions. These findings confirmed FDH as the diagnosis.
We conducted a literature search across major English (PubMed, Embase, Web of Science) and Chinese (CNKI, Wanfang Data) databases using the keywords including “familial dysalbuminemic hyperthyroxinemia,” “familial dysalbuminemic hypertriiodothyroninemia,” and their Chinese equivalents to identify relevant studies published before June 2025. Currently, large-scale clinical studies on FDH remain limited, with the existing literature predominantly comprising case reports.
To date, five pathogenic mutations associated with FDH have been identified (Table 3). Among these, the R218H/R242H mutations demonstrate the highest reported incidence. The ALB R218H/R242H mutation replaces the subdomain IIA-associated R218 residue, inducing conformational changes at the thyroxine-binding site that alleviate steric hindrance and cause abnormally increased albumin-T4 binding affinity. 12 This variant typically elevates serum TT4 to 1.1–1.8 times the upper reference limit with a mild concomitant increase in TT3 and reverse T3, consistent with our proband. 13 Although FT3 and FT4 levels in FDH patients are typically normal, the proband in this study consistently exhibited elevated FT4 levels, a finding potentially linked to the specific assay methodology used. The primary methods for measuring FT4 are direct and indirect assays. The direct method demonstrates superior accuracy by avoiding interference from binding proteins, while high cost and technical complexity typically limit its use in research settings. The one-step indirect method, which employs labeled T4 analogs competing with serum FT4 for limited antibody binding sites, is susceptible to interference from the abnormally increased albumin-binding affinity in FDH patients, potentially yielding falsely elevated FT4 results. By contrast, the two-step indirect method enhances accuracy by incorporating a wash step to minimize analog contact. 14 Zhao et al. demonstrated in a clinical study of 16 FDH patients carrying the R218H mutation that FT4 values measured using the Roche method (one-step) showed closer agreement with reference values obtained by equilibrium dialysis compared to the Beckman method (two-step). This finding suggests that inaccurate FT4 measurement in FDH patients is not solely attributable to the one-step methodology, but may also result from interference by specific buffer formulations. 15
Spectrum of reported ALB gene mutations associated with FDH.

FDH, familial dysalbuminemic hyperthyroxinemia; FT4, free T4; rT3, reverse T3; T3, triiodothyronine; T4, thyroxine; TFT, thyroid function test.
The coexistence of FDH and PTC is rare, with only three female cases reported in Chinese literature and none in English publications, making our case the first reported in a male patient.16,17 Current evidence suggests a coincidental association between FDH and PTC, as both conditions are relatively rare and appear to involve distinct pathogenic pathways. Specifically, FDH arises from ALB gene mutations that impair albumin function, whereas PTC is primarily driven by oncogenic mutations in genes such as BRAF or RAS, with no known mechanistic link to the albumin pathway. 13 Thus, the prevailing evidence supports classifying their co-occurrence as coincidental. Whether a biological link exists between FDH and PTC requires validation through more reported cases and mechanistic research.
Nevertheless, misdiagnosis of FDH as hyperthyroidism remains an important clinical issue, as demonstrated in the first reported case of a 61-year-old female. 16 The patient consequently received unnecessary antithyroid drug (ATD) treatment for years due to misdiagnosis. In patients with concurrent PTC, this approach is harmful by delaying necessary surgery and creating iatrogenic hypothyroidism. The resulting elevation in TSH stimulates the proliferation of PTC and leads to thyroid swelling and vascular congestion, thereby potentially raising the risk of intraoperative bleeding. 2 After the self-discontinuation of ATDs, the patient’s FT4 and TT4 levels remained elevated. Genetic testing identified an ALB R218H mutation, which led to the confirmation of FDH based on the characteristic thyroid abnormalities observed across three generations. The patient was later diagnosed with PTC, prompting total thyroidectomy and subsequent radioiodine (I131) remnant ablation. Her serum TSH eventually reached the suppression target, although the final L-T4 maintenance dose was not documented.
By contrast, the two cases reported by Han et al. 17 in 2023 were diagnosed with FDH following the discovery of inadequate TSH suppression after PTC surgery. A 52-year-old female did not achieve target TSH levels with L-T4 monotherapy after total thyroidectomy, whereas a 33-year-old female, although achieving adequate TSH suppression with L-T4 after subtotal thyroidectomy, developed intolerable palpitations. The ALB R218H mutation was detected in both patients, who were then effectively managed with L-T4 and DTE combination therapy, resulting in stable TSH levels without adverse effects.
The heterogeneous manifestations and therapeutic outcomes in these cases illustrate the specific complexities of managing the coexistence of FDH and PTC. Although FDH itself typically requires no intervention, its characteristic hormone profile, especially persistently elevated TT4, complicates postoperative TSH suppression. Clinicians are often hesitant to increase L-T4 dosing when TT4 levels are persistently elevated, even with suboptimal TSH levels. In our proband, TSH suppression was suboptimal with initial L-T4 monotherapy. The introduction of DTE, which supplies T3 directly, successfully achieved adequate TSH suppression. This finding is consistent with results from a randomized controlled trial (RCT) showing that combination T3/T4 therapy achieved greater TSH reduction than L-T4 monotherapy. 18 One proposed hypothesis suggests that exogenous T3 might achieve more effective central feedback than endogenously converted T3. It has also been speculated that in FDH, the high-affinity binding of T4 to mutant albumin could potentially impede free hormone release, whereas T3 binding is less affected. 19 Thus, it is plausible that combination therapy with L-T4 and DTE might help circumvent the delayed tissue delivery of T4, thereby contributing to TSH suppression via the negative feedback mechanism.
However, DTE is associated with several clinical limitations. Its supraphysiological T3:T4 ratio may produce transient serum T3 peaks, which can trigger symptoms such as palpitations, anxiety, and sweating.20,21 Significant batch-to-batch variability in its hormone content compromises dosing consistency and long-term stability. 20 Furthermore, support for DTE is constrained by limited evidence, as highlighted in a 2024 review that criticized the generally low quality of existing studies. 22 While theoretical concerns regarding long-term cardiovascular and bone safety persist, clinical data remain inconsistent, as illustrated by a 2024 meta-analysis of 16 RCT found no significant difference in resting heart rate between DTE and L-T4 regimens. 23 Monitoring is also challenging due to T3 fluctuations and the absence of a standardized target range for FT3. 21 Thus, more high-quality research is necessary to clarify the efficacy and safety of L-T4 and DTE combination therapy.
Conclusion
In conclusion, we report a patient who maintained normal TSH levels with L-T4 monotherapy after PTC surgery yet exhibited persistently elevated TT4 and FT4. Genetic testing confirmed FDH. Ultimately, combination therapy with L-T4 and DTE achieved target TSH suppression. This case underscores that persistent biochemical discordance (elevated T4 with non-suppressed TSH) after thyroidectomy necessitates consideration of rare binding disorders such as FDH. Molecular confirmation is recommended before therapeutic modification to avoid inappropriate dose reduction.