
Abstract
Objective:
Recent guidelines about Non-classical Congenital Adrenal Hyperplasia (NCCAH) indicate that if the peak cortisol response to the adrenocorticotropic hormone (ACTH) stimulation test is <14–18 µg/dL (monoclonal/polyclonal assay), it may be essential to augment the hydrocortisone dosage during significant stress events. In the current setting, when genetic diagnoses are increasingly widespread in NCCAH, there is a paucity of studies concerning cortisol responses to prevalent mutations.
Aims:
Our objectives are to investigate the outcomes of ACTH stimulation tests in children with NCCAH, delineate the biallelic mutations associated with insufficient cortisol responses, and evaluate the frequency of cortisol insufficiency in individuals with the V281L genotype. Our secondary objective is to assess the necessity of the ACTH stimulation test based on these results.
Methods:
Reviewing the retrospective medical records of 45 individuals diagnosed with NCCAH based on exacerbated clinical and biochemical findings, the mean age was 9.8 years (range: 2.4–17.9 years). Study participants had stimulated 17-hydroxyprogesterone (17-OHP) >10 ng/dL and had been investigated for 11 common CYP21A2 mutations. A peak cortisol response below 18 µg/dL was insufficient for our polyclonal assay. The cases were divided into two groups: Inadequate/low cortisol response group (LCR; n: 11) and adequate/normal response group (n: 25). We correlated biallelic genotype and cortisol response. Genotypes were classified as mild/mild (M/M), mild/severe (M/S), and heterozygous on a single allele based on the genotype’s effect on enzyme activity. P30L, V281L, P453S, and R339H are mild NCCAH mutations, whereas Q318X, R356W, exon 6 cluster mutations, Intron2A, and 8 bp deletions were severe. All cohorts with the V281L variant were separated into three groups (M/M, M/S, mild/undefined) and evaluated separately.
Results:
Almost one-third of patients had inadequate cortisol reserve. In the LCR group, 17-OHP 0’ levels were greater, but cortisol 0’ levels were lower, with no difference in stimulated responses (15.4 ± 10.5 vs 7.8 ± 6.2, p: 0.016 for basal 17-OHP; 40.7 ± 48.6 vs 32.3 ± 26.4, p: 0.63 for stimulated 17-OHP). The 17-OHP cutoff values were found to be 4.2 and 30.4 ng/dL, with 100% specificity for both groups. In 36% of the LCR group, mild–mild mutations were found. ACTH, cortisol (0’ and peak), and 17-OHP (0’ and peak) levels were similar in mild/mild and mild/severe groups. The V281L mutation was present in 86.1% of our cohort, with inadequate cortisol response in 29% of cases with V281L mutation. Differences were observed between the 0’ and peak cortisol levels in the V281L groups, with post hoc analysis indicating a difference between one allele heterozygous (mild/undefined) and mild/severe group.
Conclusion:
Our findings emphasize that a significant portion of patients with the mild/mild V281 mutation exhibit inadequate cortisol responses, highlighting the importance of the ACTH stimulation test in diagnosis and the need for careful monitoring in clinical treatment, as well as consideration of genotype variations.
Introduction
Congenital adrenal hyperplasia (CAH) is one of the most common inherited endocrine disorders characterized by genetic mutations in the enzymes that produce steroid hormones such as cortisol, aldosterone, and androgens. 1 Insufficient activity of 21-hydroxylase (CYP21A2 mutation), necessary for converting 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol, is the primary cause of CAH. Due to the loss of 21-hydroxylase function, three major phenotypes are observed under two main categories: Classical CAH (including Salt-Wasting (75%) and Simple Virilizing forms (25%)) and Nonclassical CAH (NCCAH). 2 While classical forms are recognized by severe adrenal insufficiency (AI), salt-wasting crises, and significant virilization in neonates and early childhood, NCCAH presents as a milder, late-onset form that can be symptomatic with varying degrees of postnatal virilization during childhood through to young adulthood or even sometimes asymptomatic.3,4
While lifelong glucocorticoid therapy is required for survival in classical CAH, treatment for NCCAH is indicated for progressive symptoms of hyperandrogenism, not for cortisol sufficiency. Unlike classical forms, increased adrenocorticotropic hormone (ACTH) levels are not typically observed in NCCAH; intra-adrenal cortisol secretion is generally normal or near-normal.5–8 Moreover, while the risk of AI is generally considered less pronounced in NCCAH, several studies have addressed this issue to varying extents.9–13 Although it is widely believed that a modest decrease in enzyme activity (20%–50%) is sufficient to maintain cortisol secretion, there are still conflicting views on the adequacy of cortisol response in these patients.9,10–14 Several studies have reported an insufficient cortisol response to ACTH stimulation in a significant number of individuals with NCCAH. Indeed, studies have shown that the standard dose ACTH stimulation test, regarded as the gold standard diagnostic tool, can result in subnormal cortisol responses in 30%–60% of cases,10–15 and treatment recommendations have been made even for asymptomatic cases. Nevertheless, the clinical relevance and long-term implications of these findings are not yet fully established, and treatment decisions—especially for asymptomatic patients—remain controversial. Although most patients with NCCAH and inadequate cortisol reserve do not have symptoms of AI, the fact that there are cases whose nonspecific symptoms improve has brought treatment recommendations to the agenda.15,16
Mutations in CYP21A2, a gene on chromosome 6’s short arm in the HLA class III area, result in a deficiency of the steroid 21-hydroxylase enzyme. Over 300 mutations have been identified in the CYP21A2 gene. Approximately 65%–75% of patients with 21-hydroxylase deficiency are compound heterozygotes, carrying two different pathogenic variants on each allele. In individuals with a compound heterozygous genotype, the clinical phenotype is typically determined by the milder of the two mutations. Mild variants, such as V281L and P30L, have been shown to retain 20%–50% of normal enzymatic activity, whereas severe variants—such as large deletions, gene conversions, exon 6 cluster mutations, I2 splice, Q318X, and I172N—result in enzyme activity levels of 0%–2%.17–19 There is a generally strong genotype-phenotype correlation, and individuals carrying at least one mild mutation typically present with NCCAH. 18 The V281L mutation is among the most common genetic variants associated with NCCAH. The prevalence of NCCAH varies by ethnicity and is estimated to be approximately 0.1% in the general population, increasing to 1%–2% in Hispanics and Yugoslavs, and up to 3.7%–4% in Ashkenazi Jews in earlier reports.3,20 In a Turkish study based on genetic analysis of children presenting with signs of androgen excess, the prevalence of NCCAH was found to be 4.7%, a rate comparable to those earlier reported in Ashkenazi Jewish populations.20,21 These findings suggest that the prevalence of NCCAH is relatively high in Turkey and raise the possibility that the V281L mutation may represent a founder mutation in this population. However, more recent data indicate that the prevalence in Ashkenazi Jews may be lower than previously thought, with a recent study reporting a prevalence of 0.5%. 22 Furthermore, there is currently no nationwide study reflecting the general population carrier frequency or an updated prevalence of NCCAH in Turkey. While the V281L mutation is expected to present as an NCCAH phenotype in both homozygous and compound heterozygous genotypes, it may present with clinical features in 2.8% of cases as a classical form. Although there is a genotypic correlation with the CAH phenotype, variations can be observed.17–19
While the relationship between 21-hydroxylase mutations and adrenal function has been previously investigated, data focusing specifically on cortisol reserve in individuals carrying the V281L variant remain limited. In parallel, the role of genetic testing in the diagnostic approach to CAH has become increasingly prominent in recent years. According to the Endocrine Society clinical practice guidelines, the diagnosis of CAH is primarily based on biochemical evaluation, particularly elevated baseline or stimulated 17-OHP levels. Genetic testing plays a complementary role in confirming the diagnosis, predicting the phenotype, guiding treatment, and providing genetic counseling. However, in clinical settings, due to the difficulty in accessing the synthetic ACTH (Synacthen 250 mcg), the invasive nature of the stimulation test, and the increased availability and reliability of molecular testing, CYP21A2 mutation analysis is now more commonly used in many centers to support the diagnostic process.
This study aims to evaluate the basal and stimulated cortisol response of standard-dose ACTH stimulation tests in children with NCCAH, identify the genotypic characteristics of patients with inadequate cortisol responses, and evaluate cortisol reserve in patients with NCCAH, particularly those carrying the V281L variant. The findings will shed light on the necessity of ACTH stimulation testing in diagnosing and help develop more effective treatment strategies for managing the disease.
Methods
The medical records of 45 genetically confirmed NCCAH patients (37 girls, 7 boys) followed up at Ankara University Pediatric Endocrinology Department between January 2015 and December 2022 were retrospectively analyzed to retrieve data about the clinical presentation, CYP21A2 molecular analysis, and hormone levels on the ACTH stimulation test.
The ethics committee obtained ethical approval under the tenets of the Declaration of Helsinki (approval number: 02-141-19).
NCCAH was diagnosed based on a basal 17-OHP level of 2 ng/mL, demonstrating a standard dose ACTH stimulation test peak 17-OHP >10 ng/mL or identifying a CYP21A2 mutation. 5
Molecular genetic analysis was conducted on all patients for mutations that account for more than 90% of the mutations causing the disease: P30L in exon 1, A/C655G in intron 2 (IVS2 (intron 2 splicing mutation)), an 8-bp deletion in exon 3, I172N in exon 4, a 3-point mutation cluster in exon 6 (I236N + V237E + M239K), V281L and L307F in exon 7, Q318X and R356W in exon 8, and P453S and P482S in exon 10.
During diagnosis or follow-up, the peak cortisol and 17 OHP levels to the standard dose ACTH stimulation test (0’–30’–60’) with Synacthen 0.25 mg/m2 up to 0.25 mg were examined. Five patients were tested after a 72-h break in treatment. Menstruating female patients were tested in the early follicular phase.
ACTH and cortisol were assayed by electrochemiluminescence (COBAS e411 and e801 analyzers; Roche Elecsys II, Roche Diagnostics GmbH, Mannheim, Germany.) using polyclonal antibodies. The intra- and interassay coefficients of variation were between 1.6%–4.4% and 1.3%–4.2%, respectively. A stimulated cortisol level below 18 µg/dL was considered suggestive of AI by assay characteristics and clinical guidelines. 5 17-OHP concentration was measured using a radioimmunoassay (DIA Source Immunoassay SA, Belgium, DIA Source Immunoassay SA, Louvain-la-Neuve, Belgium).
Patients were divided into two groups based on stimulated peak cortisol levels: the insufficient/low cortisol response group, with a peak cortisol response below 18 µg/dL (500 nmol/L), and the adequate cortisol group, with a response above 18 µg/dL. A peak cortisol response below 14 µg/dL was defined as severe cortisol deficiency. ACTH stimulation test responses, genetic analysis results, and clinical features were compared between the low and normal cortisol response groups.
The evaluation of genetic results was carried out by dividing the patients into three groups based on the degree of predicted enzymatic compromise 18 : those with biallelic nonclassic mutations (mild/mild), those with compound heterozygosity for one mild (20%–60% residual activity) and one severe/classic mutation (2%–5% residual activity; mild/severe); and those with a single mutation (heterozygote; mild-severe/undefined). P30L, V281L, P453S are mild NCCAH mutations, whereas Q318X, R356W, Exon 6 cluster, Intron2A (IVS2), and 8 bp deletions were severe.3,23 In this study, cases with a single heterozygous mutation but diagnosed with NCCAH due to clinical presentation and peak 17-OHP >10 ng/mL were evaluated separately from the evaluation of cortisol response through genetic classification, as it could not be clarified with further sequencing methods whether the mutation in the second allele was mild or severe.
The study did not include patients with classical (salt-wasting type, simple virilizing type) CAH or carriers.
Patients carrying the V281L variant were categorized into three genotype groups (mild/mild, mild/severe, and mild/undefined) and were evaluated separately in the statistical analyses.
Statistical analysis
Statistical analyses were performed using SPSS 19.0 (IBM SPSS Statistics; IBM software, IBM SPSS Statistics, IBM Corp., Armonk, NY, USA). For comparisons between groups, the Student’s t test was utilized for normally distributed data, while the Mann–Whitney U test was employed for non-normally distributed data. To assess differences among multiple groups, the Kruskal–Wallis test was conducted, followed by post hoc analysis using Dunn’s test and Bonferroni correction to identify specific group differences.
Receiver operating characteristic (ROC) analysis was performed to evaluate the diagnostic performance of the measured parameters, and the area under the curve (AUC) was calculated to determine the accuracy of the tests.
Statistical significance was defined as p < 0.05 for all analyses.
Results
Basal characteristics of patients
Forty-five patients diagnosed with NCCAH were included in this study. The average age was 9.8 ± 3.7 years (2.4–17.9 years), with 84.1% girls (n = 37). The most common presenting symptoms were premature pubarche (72.7%), followed by hirsutism (15.9%) and family screening (11.3%). All cases required hydrocortisone treatment except four cases diagnosed through screening.
The most frequently observed genotype was compound heterozygosity (39.4%), followed by homozygous genotypes (28.9%) and heterozygous genotypes (31.5%). Nine patients, diagnosed based on elevated 17-OHP levels and genetic evaluation, who could not be recalled for the ACTH stimulation test, were excluded from the study.
While basal and stimulated cortisol levels in the study group ranged from 4.7 to 35.4 and 6.1 to 49 µg/dL, respectively, 30.5% of them had an inadequate cortisol response to the ACTH stimulation test. The patients are subdivided into two groups according to the stimulated cortisol response. Twenty-five had an adequate peak cortisol response, and 11 had an insufficient response (Table 1).
Comparison of clinical parameters according to peak cortisol response in 36 patients undergoing ACTH stimulation test.
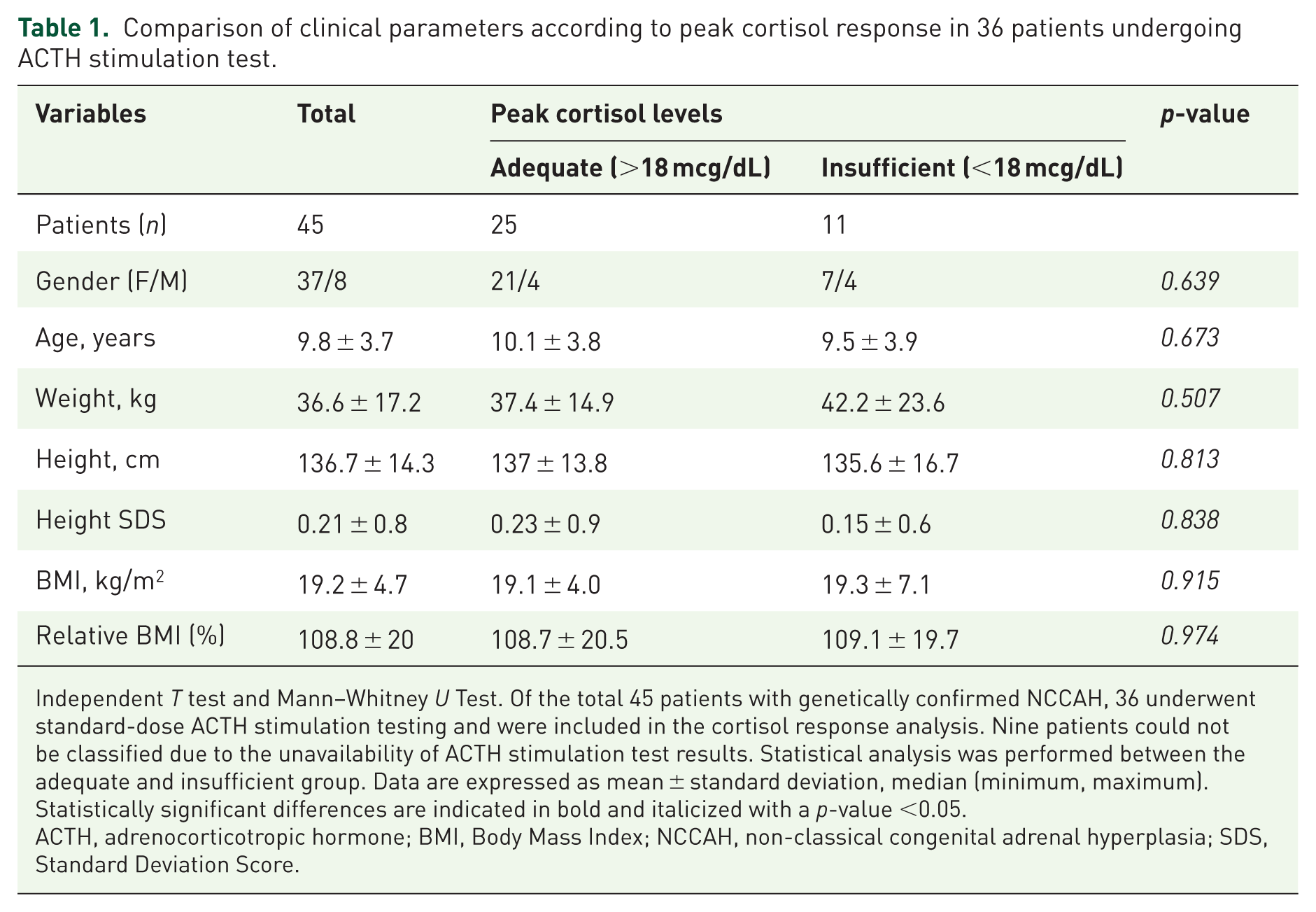
Independent T test and Mann–Whitney U Test. Of the total 45 patients with genetically confirmed NCCAH, 36 underwent standard-dose ACTH stimulation testing and were included in the cortisol response analysis. Nine patients could not be classified due to the unavailability of ACTH stimulation test results. Statistical analysis was performed between the adequate and insufficient group. Data are expressed as mean ± standard deviation, median (minimum, maximum). Statistically significant differences are indicated in bold and italicized with a p-value <0.05.
ACTH, adrenocorticotropic hormone; BMI, Body Mass Index; NCCAH, non-classical congenital adrenal hyperplasia; SDS, Standard Deviation Score.
Two groups of laboratory and genetic characteristics
In the insufficient cortisol response group, 4 patients had a mild/mild genotype (homozygous V281L), and 6 patients had mild/severe genotypes (compound heterozygous: V281L/R356W (1), IVS2/IVS2 (1), V281L/del8bp (1), V281L/Q318X (2)) and 2 patients had heterozygous genotype: V281L/undefined (1), del8/undefined (1).
In the adequate cortisol response group, 7 patients had a mild/mild genotype (homozygous V281L (6), homozygous P30L (1)), and 8 patients had mild/severe genotypes (IVS2/P30L (1), IVS2/IVS2 (2), V281L/IVS2 (2), V281L/Q318X (3)) and 10 patients had a monoallelic V281L variant (heterozygous; mild/undefined; Figure 1).
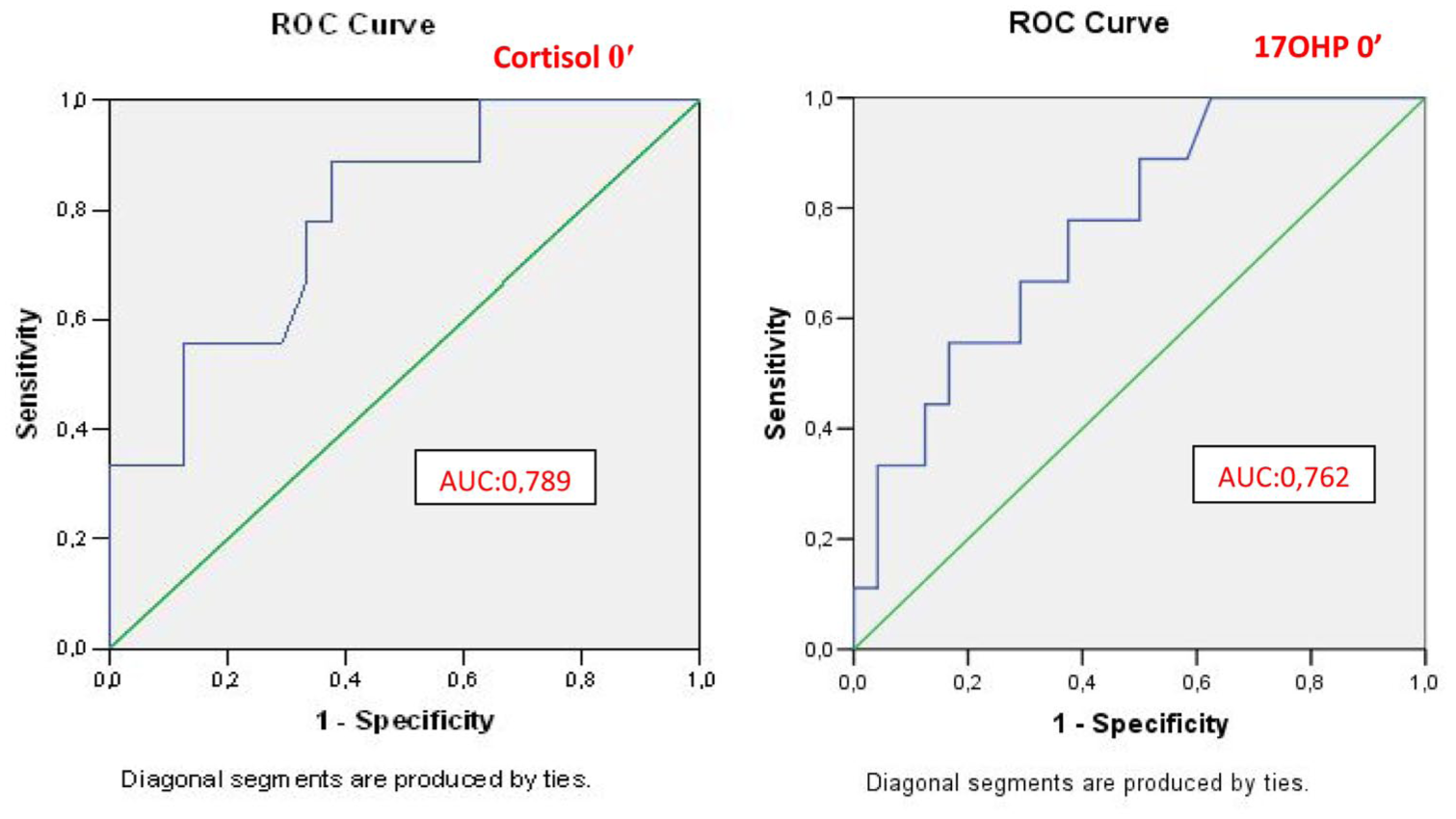
ROC analysis for basal cortisol and basal 17-OHP levels to assess partial cortisol insufficiency.
There was no difference in biallelic genotype distribution between the adequate and insufficient cortisol groups (p = 0.446). Among cases with severe cortisol deficiency, two had a homozygous V281L mutation, and one had a heterozygous deletion in exon 8 detected.
The anthropometric characteristics and age of presentation between the groups were similar. As expected based on the study design, a significant difference was observed in stimulated cortisol responses between the groups. Interestingly, basal cortisol levels also differed significantly (p = 0.029), When evaluating basal cortisol levels through ROC analysis, the cutoff value for insufficient response was found to be 13.3 µg/dL (with 77% sensitivity and 66% specificity), while the value with 100% specificity was determined to be 8.9 µg/dL (Table 2).
Comparison of basal and stimulated 17-OHP and cortisol levels to ACTH stimulation test between NCCAH patients with insufficient or adequate cortisol response groups.

Independent T test and Mann–Whitney U test. Data are expressed as mean ± standard deviation, median (minimum, maximum). Peak cortisol <18 mcg/dL was defined as the insufficient cortisol group, while over 18 was defined as the adequate group. Statistically significant differences are indicated in bold and italicized with a p-value <0.05.
ACTH, adrenocorticotropic hormone; NCCAH, non-classical congenital adrenal hyperplasia; 17-OHP, 17-hydroxyprogesterone.
In the insufficient cortisol response group, basal 17-OHP levels were higher compared to the adequate response group, while no difference was observed in stimulated responses (15.4 ± 10.5 vs 7.8 ± 6.2, p: 0.016 for basal; 40.7 ± 48.6 vs 32.3 ± 26.4, p: 0.63 for stimulated). In the adequate cortisol response group, 29.4% had a 17-OHP peak response above 40 ng/mL, whereas among cases with inadequate response, 12.5% had a response above >40 ng/mL.
In ROC analysis, the best cut-off for basal 17-OHP was 8.5 ng/mL with a sensitivity of 66% and a specificity of 70%, 100% specificity cut-off value was determined as 30.4 ng/dL with AUC 0.762 for determining insufficient response, and 4.2 ng/mL for adequate response (%100 specificity; Figure 2).

Genotype-cortisol deficiency distribution in NCCAH with V281L mutation.
Frequent mutations in NCCAH
The most frequently detected mutation in all cases was the V281L point mutation, observed in 86.1% of cases. Insufficient cortisol response was observed in 29% of cases carrying the V281L mutation. V281L mutation was categorized into three groups based on mutation type and severity for analysis. Insufficient response was observed in 44.4% of individuals with homozygous V281L mutations (mild/mild) versus 33.3% of compound heterozygous (mild/severe) cases. In addition, 10% of instances with a mutant variant discovered in a single allele (mild/undefined) showed insufficient cortisol response (Figure 1).
When heterozygous cases with one allele carrying V281L mutation were excluded, no statistically significant difference between basal/stimulated cortisol levels (p = 0.12; p = 0.56, respectively), basal/stimulated 17-OHP levels (p = 0.71; p = 0.37, respectively), or ACTH concentrations (p = 0.50) between the mild/mild and mild/severe genotype groups (Table 3).
Evaluation of basal and stimulated responses in the standard dose ACTH stimulation test according to CYP21A2 genotype including V281L variant in children with NCCAH.

p < 0.016.
One-way ANOVA and Kruskal–Wallis test. Data are expressed as mean ± standard deviation. Statistically significant differences are indicated in bold and italicized with a p-value <0.05. In the post hoc analysis, the Bonferroni correction was applied to determine differences between groups, and each comparison’s p-values were compared against the adjusted significance threshold. Compared with the heterozygote group in post hoc analysis.
ACTH, adrenocorticotropic hormone; NCCAH, non-classical congenital adrenal hyperplasia; 17-OHP, 17-hydroxyprogesterone.
When the three groups were evaluated together, significant differences were found in the cortisol 0’ (p = 0.011) and peak cortisol levels (p = 0.004), while no differences were observed in ACTH, 17-OHP 0’, and peak 17-OHP levels (p = 0.054, p = 0.61, and p = 0.08, respectively). Multiple comparisons were performed using post hoc analysis with Dunn’s procedure and Bonferroni adjustment. This post hoc analysis revealed statistically significant differences in the median cortisol scores 0’ between the mild/severe genotype and heterozygous genotype (p: 0.007, 11.03 ± 2.9 and 20.9 ± 10.2, respectively). A similar difference was also observed in the peak cortisol response (p: 0.011, 18.3 ± 7.1 and 28.9 ± 8.8, respectively).
In instances where the V281L mutation was identified in at least one allele, the stimulated 17-OHP level exceeded 40 ng/dL in seven cases and above 100 ng/dL in three cases.
Discussion
In our study evaluating the cortisol response to the standard dose ACTH stimulation test and its relationship with genetic mutations in NCCAH, we found insufficient cortisol secretion in 30.5% of our cohort and even a severe deficiency with a response below 14 µg/dL in 8% of cases. The cutoff value for 17-OHP has been determined to indicate an insufficient cortisol response above 30.4 ng/mL with 100% specificity, while a value below 4.2 ng/mL indicates sufficient response. Previous studies have reported subnormal cortisol responses in NCCAH ranging from 15% to 60%.10–16 Cortisol deficiency is typically assessed through stimulated cortisol levels, unaffected by the time of day the test is conducted. 24
We noted significant differences between peak and basal cortisol levels, with the adequate response group showing a basal level of 16.8 ± 9.5 (p: 0.02). The optimal measurement time is suggested to be before 09:00, with a cutoff value of 375 nmol/L (13.5 μg/dL).25,26 Due to nonspecific clinical findings and challenges in obtaining necessary medications for the dynamic test, especially in our country, clinicians often rely on basal cortisol and ACTH levels for diagnosing AI. 27
While various cortisol levels are proposed, a basal cortisol level <100 nmol/L (3 μg/dL) or >330 nmol/L (12 μg/dL) indicates that dynamic testing is unnecessary. 28 A study indicated that a basal morning cortisol level of ⩾400 nmol/L (14.4 μg/dL) is indicative of an adequate response to the ACTH stimulation test. 29 In our study, we determined the cutoff for insufficient response in NCCAH cases to be 13.3 µg/dL (with 77% sensitivity and 66% specificity). In comparison, the value with 100% specificity was found to be 8.9 µg/dL. Studies showing inadequate responses in the ACTH stimulation test reported sufficient basal cortisol levels. Weintrob et al. 11 reported normal basal levels in children with NCCAH, while the delta cortisol response and peak levels were significantly lower compared to controls, suggesting that basal cortisol levels may be misleading in this population. Basal cortisol sensitivity for diagnosing AI is 60%. 30 In nonspecific symptoms where basal tests do not assist in diagnosis or instances of subclinical AI, dynamic tests should be used to support diagnosis by evaluating the response under stress conditions. 31 It has been noted that in NCCAH, cases may achieve cortisol normalization with a delayed response observed 48–72 h after ACTH stimulation. 32 Huerta et al. 9 suggested that in patients, exaggerated 11-deoxycortisol production due to adrenal cortical hypersensitivity partially accounts for preserved cortisol production in 21-OH-deficient NCCAH. However, when evaluating adrenal medullary functions across all subtypes of adrenal hyperplasia requiring treatment, including NCCAH, impairments in epinephrine and cortisol secretion were identified. 14 Presentation with clinical symptoms has been reported in cortisol-deficient NCCAH. In the ACTH stimulation test, attention should be paid to the study methods and new cutoff limits. Considering the safety profiles and accuracies, corticotropin analogs such as tetracosactrin (Synacthen®), (tetracosactrin): Novartis Pharma AG, Basel, Switzerland or (Cortrosyn®) (cosyntropin): Amphastar Pharmaceuticals, Inc., Rancho Cucamonga, CA, USA are used as first-line stimulation tests, with a cutoff value recommended at 18 µg/dL. This value has been obtained using existing cortisol tests, particularly the widely used Elecsys® Cortisol immunoassay. For newer assays, such as those using LC-MS/MS, a competitive immunological test that employs monoclonal rather than polyclonal antibodies, it is suggested that a peak cutoff value of 13.5 µg/dL be used in assays with different standardizations (e.g., Elecsys Cortisol II). 33
In our center, due to the recent introduction of new immunoassays, we used a cutoff value of 18 µg/dL in our retrospective study. In assessing AI, attention should be paid to the new cutoff values. A few studies indicate insufficient response to the ACTH stimulation test in NCCAH.10–15,34 Ghizzoni et al. 34 found that 28% of NCCAH cases had responses below 18.2 µg/dL. In another study evaluating 161 patients, a low response of less than 15 µg/dL was obtained in 32% of cases. Stoupa et al. 16 reported that despite a basal cortisol level of 12.9 µg/dL in 35 patients, 61% had responses below 18 µg/dL during the High-Dose Synacthen Test (HDST) and noted severe symptoms of fatigue and weakness that improved with hydrocortisone treatment in two cases. Karachaliou et al. 10 found a 21.2% insufficient cortisol response in 31 NCCAH cases without performing mutation typing. In our study, despite a median basal cortisol level of 12.8 µg/dL in all cases, 30.5% showed responses below 18 µg/dL in the ACTH stimulation test, suggesting a risk of subclinical AI.
An interesting observation regarding the cases with insufficient cortisol levels reported in the literature is that many of them had a compound heterozygous genotype, including one severe mutation.8,13,17,24 This is probably related to the fact that most of the mutations identified in CAH are compound heterozygous (65%–75%). Since the enzyme activity determines the phenotype in compound heterozygous cases, it is expected to be seen clinically as a mild mutation. 35 In the limited number of evaluations conducted on mutation type and cortisol response, Weintrob et al. 36 found similar basal and stimulated cortisol levels in mild/mild and mild/severe genotype groups. Other studies express differing views. Koren et al. 25 reported that in their large cohort of 122 patients, the majority had the V281L mutation and that in the mild/severe group, failure of the ACTH stimulation test (75%) was more prevalent than in the mild/mild group (39%). Nandagopal et al. 13 identified a compound heterozygous mutation, one of which was a severe mutation, in 10 families. Three cases showed subnormal cortisol responses when subjected to a standard dose ACTH stimulation test. Our study did not find a difference in cortisol response between the two genotypes, which may be due to the smaller sample size.
When examining the commonly seen mild V281L mutation, we found that half of the cases in the cortisol-deficient group had the mild/mild V281L mutation, while one case had a one-allele heterozygous mutation. The patient with the heterozygous mutation was a 14-year-old who presented with hirsutism; their peak cortisol response was 16.2 µg/dL, and the peak 17-OHP response was 12.7 µg/dL. The stimulated cortisol responses in compound heterozygous cases and homozygous V281L cases were similar. A significant difference in basal and stimulated cortisol levels was shown between heterozygous and mild/severe mutation groups. Other studies reporting insufficient cortisol response in NCCAH cases have also presented similar findings related to genotype.10,18,35–38
Among the approximately 300 mutations identified in the 21-hydroxylase gene, the V281L point mutation occurring in exon 7 is the most frequently detected mutation in NCCAH cases. In clinical practice, NCCAH can occur in 2.6% of cases, sometimes presenting with a simple virilizing phenotype. In compound heterozygous cases, clinical presentation is expected to reflect the enzyme activity associated with mild mutations. However, while the presence of a mild mutation may alleviate clinical presentation, it does not ensure sufficient cortisol response.
Therefore, patients with NCCAH and suboptimal cortisol response may exhibit signs of AI (often severe fatigue and low blood pressure) during periods of stress, even in the absence of treatment or after glucocorticoid therapy is discontinued in adulthood. Unlike classical CAH, treatment is not indicated for life. However, the frequency of inadequate responses, delayed cortisol responses, nocturnal cortisol deficiency, and symptomatic presentations in individuals have raised concerns regarding the adequacy of cortisol response during stress.15,16,25
As such, even if basal cortisol and ACTH levels are sufficient, stress response should be evaluated. It has been suggested that if a stimulated cortisol level is present at 500 nmol/L (or lower in more specific recent immunological tests), the continuation of glucocorticoid therapy or stress dosing during significant stress situations may be necessary. The 2018 Endocrine Society guidelines indicate that for previously untreated NCCAH patients, stress replacement medication is not advised under extreme stress if there is no cortisol response below normal after diagnostic cosyntropin stimulation. 5
In our cohort, the basal 17-OHP levels were significantly higher in the insufficient response group, while there were no differences in stimulated 17-OHP levels. These results are consistent with other studies investigating genotype, cortisol, and 17-OHP responses.10,35–38 Dörr et al. 37 found cortisol deficiency in 27.4% of their cohort, 55% of whom had a stimulation test, and showed that both basal and stimulated 17-OHP were higher in the severe-mild mutation group, although not statistically significant. Another study reported higher basal and stimulated 17-OHP levels and lower cortisol levels in the mild/severe genotype group compared to the mild/mild genotype group, with a notable difference only in basal 17-OHP levels. 38 Koren et al. 25 (122 NCCAH subjects, median age 9.0 years) reported that ACTH stimulation test failure occurred in both genotype groups, but the rate of failure was significantly higher in the mild/severe group (75%) compared to the mild/mild group (39%). The data indicate that elevated basal 17-OHP levels correlate with an increased probability of inadequate cortisol response, necessitating careful examination irrespective of genotype.
A strength of our study is that it is among the limited number of studies investigating the association between specific CYP21A2 genotypes, specifically focusing on the V281L variant, and cortisol response in children with NCCAH. Although the number of cases is relatively small, all patients were evaluated at a single tertiary center using standardized protocols, enhancing the reliability and internal validity of our findings. Limitations include the small sample size, the retrospective design, and the fact that formerly steroid hormone measurements via LC-MS/MS were not widely available in Turkey. Also, due to the retrospective design of the study, the rarity of the condition under investigation, and the exclusion of cases with only a single (heterozygous) mutation identified in the molecular analysis, given the autosomal recessive nature of the disease, a formal power analysis for sample size calculation was not performed.
In conclusion, even in patients with the common V281L genotype and mild/mild mutations in NCCAH, suboptimal cortisol secretion is frequently observed. To clarify its clinical significance, prospective studies should comprehensively record clinical symptoms. Additionally, the standard dose ACTH test should remain the gold standard for cases diagnosed through genetic analysis. Careful attention is needed regarding stress coverage and/or the continuation of glucocorticoid therapy into adulthood for these subjects.