
Abstract
Background:
Diabetic foot ulcer (DFU) is a common and highly morbid complication of diabetes with high unmet medical needs. AUP1602-C, a topical four-in-one gene therapy medicinal product (GTMP), consisting of a Lactococcus cremoris strain that produces fibroblast growth factor-2, interleukin-4, and colony-stimulating factor-1, is a promising novel treatment for DFU.
Objectives:
The aim of this first-in-human study was to investigate whether AUP1602-C is safe and effective in improving wound healing and quality of life (QoL) in patients with non-healing DFU (nhDFU), and to determine the recommended phase II dose.
Design:
Phase I, single-arm, open-label, uncontrolled, dose escalation study.
Methods:
The study consisted of four cohorts of patients receiving AUP1602-C as a single dose of 2.5 × 105 colony-forming units (CFU)/cm2 ulcer size or as repeated doses between 2.5 × 106 and 2.5 × 108 CFU/cm2 administered 3 times per week for 6 weeks. Within each cohort, a 3 + 3 scheme for monitoring safety, tolerability, and efficacy was applied.
Results:
In total, 16 patients aged 53–80 years were included, 3 each in the safety and low dose, 4 in the medium dose, and 6 in the high-dose cohort. AUP1602-C demonstrated a favorable safety profile with almost 100% dosing compliance. The most frequently reported side effect related to treatment was skin maceration. No serious adverse reactions, systemic toxicity, deaths, or side effects suggestive of immunogenicity, hypersensitivity, allergic reaction, or dose-limiting toxicities related to treatment were reported. No biodistribution events were observed and shedding-related events were rare and did neither show accumulation nor dose dependency. The recommended phase II dose of 2.5 × 108 CFU/cm2 demonstrated complete healing in 83% of patients without recurrence of ulcers during follow-up.
Conclusion:
AUP1602-C was safe and well tolerated and demonstrated dose-dependent efficacy in patients with nhDFU. Data supports further clinical development of AUP1602-C.
Trial registration:
The study was registered in ClinicalTrials.gov (NCT04281992) and ClinicalTrialsRegister.eu (2018-003415-22).
Keywords
Introduction
Diabetes mellitus (DM) currently affects 536.6 million people worldwide, rising to 12.2% (783.2 million) worldwide among 20- to 79-year-olds in 2045. 1 Diabetic foot ulcer (DFU) is a common and highly morbid complication of DM, with a lifetime risk of 19%–34% in diabetic patients and rising incidence. 2 Approximately 18.6 million people worldwide are affected by DFU each year. 3 Progression to ulceration, involving neuropathy, ischemia, and infections caused by minor trauma, is well established. Consequences of DFU include decline in functional status, infection, hospitalization, lower-extremity amputation, and death. Morbidity following incident ulceration is high, with recurrence rates of 60% at 3 years and 5-year mortality of 50%–70%. 2 Additionally, 10% of patients die within 1 year of their first DFU diagnosis4,5 and the risk of death for a DFU patient at 5 years is 2.5 times higher than that of a DM patient without an ulcer. 6
Approximately 20% of patients who develop DFU require lower-extremity amputation, either minor (below the ankle), major (above the ankle), or both, 7 while non-healing DFUs (nhDFU) account for more than 60% of all non-traumatic lower limb amputations. 8 DFU is a leading cause of hospitalization in DM patients, even exceeding rates of congestive heart failure, depression, or renal disease. 9
DFU results in a massive burden on health resources. In Europe, the EURODIALE study performed in 2008 showed the mean annual cost per DFU patient was €10,09110 with hospitalization being the most relevant direct cost. In the United States, the cost of DFU management ranged from $9 to $13 billion between 2007 and 2011, with DFU adding $11,710–$16,833 incremental costs to a patient’s annual healthcare, doubling the cost of providing diabetes care. 11
The current standard of care (SoC) for DFU includes glycemic control, adequate lower limb perfusion, wound debridement, infection control, moist wound care with adequate dressings, and off-loading. 12 Unfortunately, up to 70% of patients do not respond to standard treatment, 13 and the estimated recurrence rate within 1 year of ulcer healing is roughly 40%. 7 According to the national diabetic foot care audit of England and Wales by the National Health Service, approximately 48% of DFU remain unhealed after 12 weeks of treatment.14,15 Comparing the cure rates of various studies from 1999 and 2018, it was found that the 12-week rate had only slightly improved from 24% to 30%,13,16 highlighting an unmet medical need that requires fundamental new therapeutic modalities.
Current trends for advanced therapies of chronic wounds include devices or products such as negative wound pressure therapy,17,18 hyperbaric or topical oxygen therapy, 19 extracorporeal shock wave stimulation, 20 special purpose dressings,21–23 skin grafts, and bioengineered skin 24 as well as drugs or biologics, for example, locally administered growth factors. 25
A number of DFU pharmaceutical candidate products have recently failed to gain approval due to safety or efficacy reasons (e.g., HP802-247: human allogeneic fibroblasts and keratinocytes cell therapy from Smith & Nephew in 2014; Daprodustat: GSK1278863 small-molecule HIF-PH inhibitor biologic from GSK in 2015; DSC127: AT1-9R single factor peptide from Dermascience in 2015). No pharmacological wound healing product has gained market approval in both the US and EU within the last 10 years, constituting a highly unmet medical need.
The physiological process of wound healing involves a complex interplay between primarily keratinocytes, fibroblasts, endothelial cells, and recruited immune cells, and their associated extracellular matrix in a well-orchestrated cascade through hemostasis, inflammatory, proliferative, and maturation phases.26,27 However, in chronic wounds such as DFU, this tightly controlled process of wound healing is impaired and detained in one or more phases. Several pathogenic abnormalities, ranging from disease-specific intrinsic flaws in blood supply, diabetic neuropathy, angiogenesis, and matrix turnover, to extrinsic factors due to plasma cells and their related pro-inflammatory cytokines in diabetic wounds, contribute to the onset of a pro-degradative microenvironment, which results from the imbalance between matrix synthesis and degradation. Especially chronic inflammation seems to play a major role in the pathogenesis of DFU.28,29 In chronic wounds, the shift of M1-type macrophages to the alternative M2-type macrophages does not readily occur. The wound remains in a state of inflammation inhibiting the initiation of the proliferation phase and subsequent tissue regeneration.28,30
AUP1602-C consists of a Lactococcus cremoris strain, genetically engineered to produce and secrete three human therapeutic proteins, fibroblast growth factor-2, interleukin-4, and colony-stimulating factor-1, at the disease site aimed at modulating the local wound microenvironment and promoting wound healing. 31 The AUP1602-C bacterium acts as millions of bioreactors in the wound, allowing to re-start and accelerate healing of chronic ulcers by (1) awakening the immune microenvironment, (2) driving macrophage conversion from pro-inflammatory M1 to anti-inflammatory and regenerative M2 phenotype, (3) driving granulation tissue formation by increasing fibroblast proliferation and promoting angiogenesis, and (4) supporting epithelialization. The extensive non-clinical program performed with AUP1602-C demonstrated that all aspects of wound healing could be addressed and that the synergistic effect of the three produced proteins together with the intrinsic activity of the bacteria accelerates wound healing. Thus, AUP1602-C was considered to possess the desired characteristics and pre-clinically showed a favorable safety profile as prerequisite for a first-in-human (FIH) study. 31
The aim of this phase I FIH study was to investigate whether the gene therapy medicinal product (GTMP) AUP1602-C is safe and effective in improving wound healing and quality of life (QoL) in patients with nhDFU and to determine the recommended phase II dose. Thus, based on the favorable preclinical profile, an FIH study was designed to evaluate the safety, tolerability, and efficacy of AUP1602-C in DFU patients. To our knowledge, this is the first study to report positive results of a GTMP of this nature in DFU patients.
Methods
Study design
This FIH study was performed in patients with chronic nhDFU. The data presented here was collected between January 28, 2020 and March 20, 2023. This open-label, non-randomized, uncontrolled, dose-finding study was conducted at one study site in Poland (MIKOMED, IU. Łódź, Poland). Based on preclinical safety and toxicity testing as well as dose escalation studies performed in the delayed wound healing db/db mouse model, 31 the selected optimal dose for the FIH was 2.5 × 107 CFU/cm2. In the FIH study, dose escalation started at a 100-fold lower dose (2.5 × 105 CFU/cm2) for safety reasons. Finally, the study consisted of four sequentially recruited cohorts of patients who received escalating doses of AUP1602-C as follows: single administration of 2.5 × 105 colony-forming units (CFU)/cm2 ulcer size (safety dose) in Cohort 1, followed by 3 times a week repeated administration for 6–10 weeks of 2.5 × 106 CFU/cm2 (low dose), 2.5 × 107 CFU/cm2 (medium dose), or 2.5 × 108 CFU/cm2 (high dose) in Cohorts 2, 3, and 4, respectively. In the repeated administration cohorts, the actual treatment duration depended on the level of healing and treatment was discontinued once the target ulcer was completely closed or when 6 weeks of treatment were completed. Wound closure was confirmed 2 weeks later. Extension of treatment beyond 6 weeks was allowed on a case-by-case basis for a maximum of 4 additional weeks. After the last AUP1602-C treatment, all patients were followed for up to 12 months (Figure 1).

Study flowchart and treatment cycles. (a) Scheme describing the standard 3 + 3 cohort escalation process with centralized review process between the cohorts. Dashed lines highlight the actual pathway during the clinical phase I study. (b) Detailed view of dosing and treatment periods for both single administration cohort 1 as well as repeated administration cohorts 2–4.
Within each cohort, a standard 3 + 3 scheme for monitoring AUP1602-C safety and tolerability was applied. A Cohort Review Committee evaluated the safety data and made recommendations for dose escalation depending on the occurrence of dose-limiting toxicities (DLTs). Six patients were included in the recommended phase II dose cohort, irrespective of the occurrence of DLTs to keep the probability of DLTs under 20%.
The study consisted of a run-in phase, treatment phase, and follow-up periods, during which all patients received SoC as needed at the discretion of the treating physician regardless of cohort. A wound area reduction (WAR) of <30% after 2 weeks of treatment with SoC during the run-in period was used to select nhDFUs.
The primary objective of the study was to investigate the local and systemic safety and tolerability of single and repeated topical administrations of AUP1602-C in patients with nhDFU. In addition, the recommended phase II dose and treatment schedule of AUP1602-C was aimed to be determined.
Secondary objectives were to assess the efficacy of multiple administrations of AUP1602-C, and QoL. Moreover, the presence of AUP1602-C (plasmid) in the systemic circulation, urine, and feces was determined.
The study was performed in accordance with ICH Good Clinical Practice—E6 (R2) and the Declaration of Helsinki, and with approval of the study site’s responsible ethics committee (Komisja Bioetyczna Warszawski Uniwersytet Medyczny, approval no. KB/41/2019) and the Office for Registration of Medicinal Products, Medical Devices, and Biocidal Products, Poland. Written informed consent was obtained from all study participants. The trial is registered in EudraCT (2018-003415-22) and ClinicalTrials.gov (NCT04281992).
Participants
Eligible to the study were both male and female (non-pregnant and non-lactating) patients aged ⩾18 years, diagnosed with DM of type 1 or 2, having a glycosylated hemoglobin of ⩽12% and a serum creatinine level of ⩽1.5 times the upper limit of normal, and at least one target DFU of 1–9 cm2 in size that had been present for >1 month. Patients must also have either an ankle-brachial index ⩾0.7 or a toe-brachial index ⩾0.5, and toe systolic pressure of at least 50 mmHg on the foot with the target ulcer, neuropathy of the foot, and were willing and able to give written informed consent.
Main exclusion criteria were treatment with another investigational drug and/or medical device or participation in another clinical study within 2 weeks prior to start of screening/run-in period, treatment with a biologic agent, growth factors or skin equivalents within 30 days prior to start of screening/run-in period, or treatment with active wound care agents (e.g., local and systemic antibiotics or silver dressings) within 1 week prior to first study drug dosing, corticosteroids, and immunosuppressants within 2 weeks prior to first study drug dosing. Patients were also excluded if they had known hypersensitivity to any of the investigational drug or vehicle components, had an ulcer with deep abscess, or gangrene with known or suspected active infection that required antimicrobials or were positive for methicillin-resistant Staphylococcus aureus. A full list of inclusion and exclusion criteria is provided in Supplemental Table 1.
Study endpoints
The primary endpoints investigated in this study were the incidence of treatment-emergent adverse events (TEAEs) and potential DLTs. Secondary endpoints evaluated during the treatment and 12-months follow-up period were percentage of wound size reduction, proportion of patients achieving complete wound closure of target ulcer, percentage of wound volume and depth reduction, time to complete wound closure, proportion of patients with ulcer recurrence, local wound infections related to the target ulcer, local surgical procedures, amputations (minor or major) related to the target ulcer, change from baseline in health-related QoL using EuroQol-5D (EQ-5D)32,33 visual analog scale (VAS), EQ-5D utility index, and Dermatology Life Quality Index (DLQI) score, 34 change from baseline in patient’s pain intensity using a numerical rating scale, incidence of abnormal vital signs values, ECG data, echocardiogram data, ophthalmoscopy data, physical examination findings, laboratory data, assessment of biodistribution of AUP1602-C (plasmid) in blood by AUP-quantitative polymerase chain reaction (qPCR), and assessment of shedding of AUP1602-C (plasmid) in urine and feces by AUP-qPCR. From the patients who specifically consented, wound biopsies were taken for exploratory pharmacodynamic analyses on visit 1, visit 10, and follow-up visit 1.
Investigational medicinal product and reconstitution solution
The Investigational Medicinal Product (IMP) batch used in this FIH study was a monoseptic, off-white suspension for topical administration in disposable, single-use vials, manufactured according to Good Manufacturing Practice (GMP) at Wacker Biotech B.V. (Amsterdam, Netherlands). Vials contained the L. cremoris AUP1602-C drug product strain (2 mL of 8 × 1010 CFU/mL), glycerol, and sodium
The reconstitution solution was a sterile hypertonic solution containing 5% dextrose, 2.5% sodium chloride, and 1.6% sodium acetate in sterile water. The batch of reconstitution solution used in this FIH study was GMP manufactured as a sterile liquid at Eurofins PROXY (Leiden, Netherlands). Prior to clinical administration, the AUP1602-C drug product was reconstituted and serially diluted with reconstitution solution to achieve the target dose and then applied directly topically to the patient’s wound at a dose volume of 50 µL/cm2. All treatment administrations and wound care measures were done by a protocol-trained medical expert, that is, a medical doctor, podiatrist, or nurse.
The IMP and reconstitution solution were packaged into clinical kits, each containing doses for 1 week of treatment (3 + 3 vials, enough for three treatments). Clinical kits were packaged, labeled, and released for clinical use by PCI (Bridgend, UK and Milmount, Irland).
Planimetry
Wound size was measured using a LifeViz Micro (QuantifiCare, Biot France) 3D camera recording stereo images which were reconstructed into 3D images for the analysis of wound area, depth, and volume. Planimetry data was collected at each visit and used for both dosing volume determination and wound area/closure assessment. Complete wound closure of the target ulcer was defined as 95%–100% re-epithelialization with no drainage (i.e., no detectable exudate) or dressing requirement. Complete wound closure was assessed using wound size measurements by both the investigator and independent central observer and confirmed at two consecutive study visits, 2 weeks apart. Both pre- and post-debridement wound images were taken, but only the post-debridement images were used for efficacy assessments.
Standard of care
Irrespective of the cohort, all patients received SoC for DFU according to the most up-to-date international guidelines 35 during the run-in, treatment, and follow-up periods of the study. SoC comprised standard treatment for diabetes, sharp/surgical wound debridement, wound dressing for moist wound care, off-loading footwear, control of wound infections, and vascular assessments and interventions, if needed. Semipermeable transparent film dressing was used as the primary wound dressing along with a secondary foam dressing during the IMP treatment period. Dressing changes were performed with the IMP treatment. During the run-in and post-treatment follow-up periods, foam dressing was used as the primary wound dressing and changes were performed at the discretion of the investigator. Removable ankle-high off-loading footwear was provided to all patients. Patient-reported compliance with off-loading footwear was assessed from screening up to the last follow-up visit.
Biodistribution and shedding
For biodistribution and shedding purposes, an AUP1602-C (plasmid) specific qPCR method was developed and validated at IMGM Laboratories (Martinsried, Germany) to detect and quantify positive blood, urine, and feces samples. Samples were collected at multiple time points throughout the study period. Blood, urine, and feces were sampled from all cohorts prior to the first administration, on Days 2, 3, 15, and 40 (not from the safety cohort on Days 15 and 40), in week 1 and at 1, 3, and 6 months. In addition, blood was sampled on Day 1 post-dose at several time points from safety cohort (1, 2, 4, 8, and 24 h) or only 1 h after dosing for the remaining cohorts. Blood samples were used to evaluate biodistribution of AUP1602-C (plasmid) in the systemic circulation, whereas urine and feces samples were analyzed for shedding of AUP1602-C (plasmid).
Quality of life
Patient-reported outcome questionnaire was applied to determine the effect of AUP1602-C treatment on patient’s QoL. Cohort 1—single dose—was used as surrogate control, compared to Cohort 4. Baseline was compared to end-of-study values (FU-V6) using EQ-5D-5L, 33 VAS and utility index (DLQI), 34 and pain intensity perception on a 0–10 scale (no pain to worst imaginable pain). EQ-5D-5L utility index score ranges from 0 (worst health state) to 1 (best health state). EQ-5D-5L VAS is a VAS with values between 0 (worst imaginable health) and 100 (best imaginable health), on which patients provide a global assessment of their health. DLQI score ranges from 0 (best QoL) to 30 (worse QoL), with the following pre-defined score categories comparing baseline versus end of study: 0–1 (no effect at all on patient’s life), 2–5 (small effect), 6–10 (moderate effect), 11–20 (very large effect), 21–30 (extremely large effect).
Statistical considerations
No technical sample size calculation was performed for the study, nor were there formal hypotheses formulated. All inferential statistics that were used for cohort differences were considered exploratory and only of descriptive nature.
No imputation of missing values was done. All analyses were conducted on observed data. Data were summarized by means of descriptive statistics.
Patient populations were divided into two analyzable sets according to the statistical analysis plan: safety set and evaluable set. The safety set consisted of patients who received at least one dose of AUP1602-C regardless of whether they completed the study or not. All safety outcomes were assessed using the safety set. The evaluable set consisted of patients who received at least one dose of AUP1602-C and were evaluable for DLT assessment, which was defined as patients who received ⩾75% of the planned treatments during the first 4 weeks of treatment. All efficacy outcomes were assessed using the evaluable set.
Results
Study population
In total, 26 patients were screened for inclusion in the study and 16 of them were eligible and received at least 1 dose of AUP1602-C, constituting the safety set with n = 3 in Cohorts 1 and 2, n = 4 in Cohort 3, and n = 6 in Cohort 4. Fifteen out of the 16 patients of the safety set were evaluable for DLT assessment, constituting the evaluable set. One patient in Cohort 3 was excluded from the evaluable set as he/she did not receive a sufficient number of AUP1602-C treatments (⩾75% of planned AUP1602-C treatments during first 4 weeks) to be eligible for DLT evaluation and was replaced. None of the patients of the safety set withdrew from the study up to the 3-month follow-up visit. Two patients discontinued study participation after the 3-months follow-up visit, that is, one patient died of a COVID-19 infection and another patient discontinued on own decision but completed the 6-month and 12-month visits ahead of the scheduled date as discharge procedures.
For patient distribution throughout the different cohorts see Figure 2. Patient demographic characteristics, including smoking habits, a summary of diabetic history and complications, and selected baseline target ulcer characteristics are given for the safety set on a per cohort basis in Table 1.
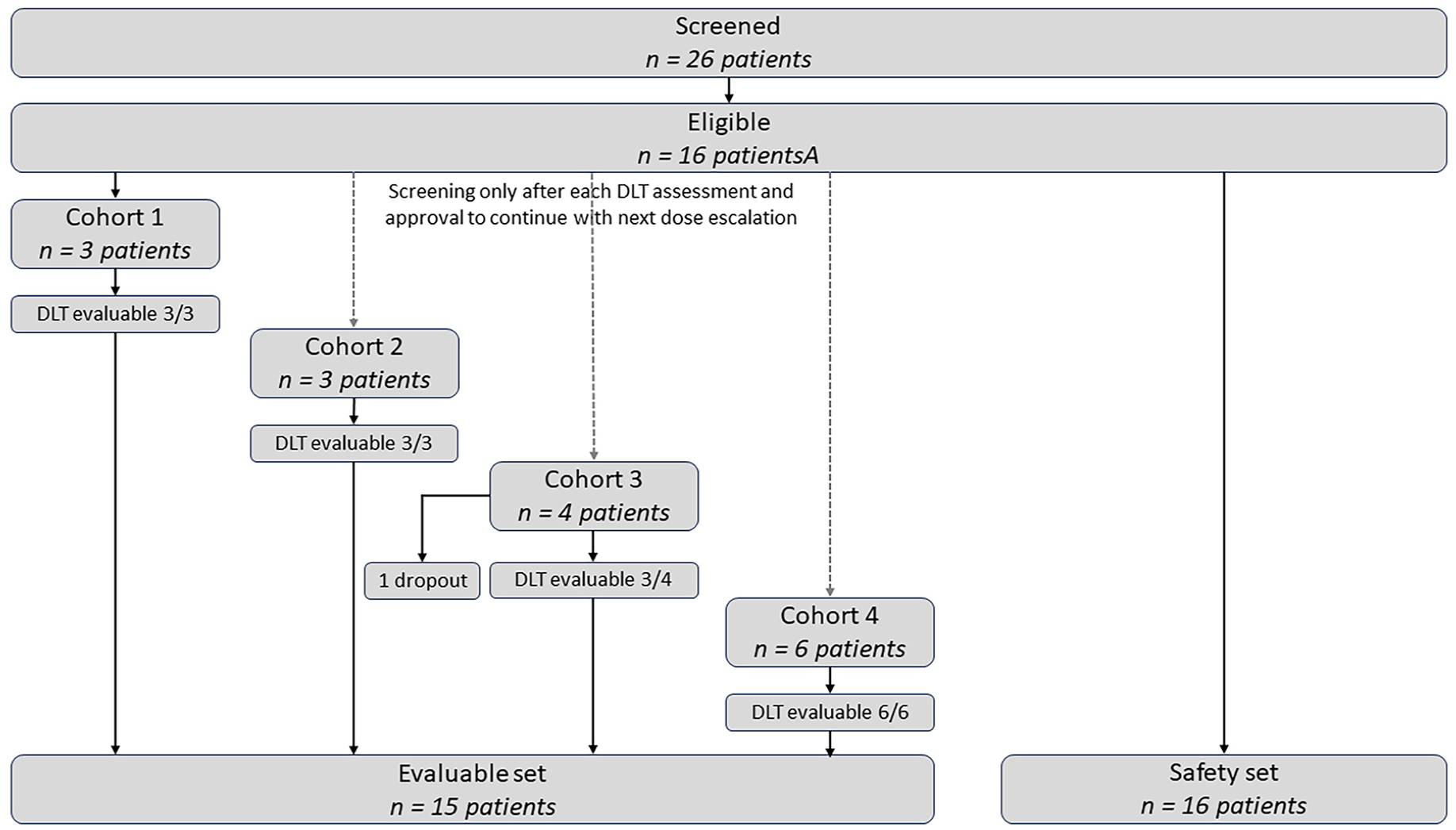
Consort chart detailing the number of screened and eligible patients as well as the evaluable and safety sets used in the statistical analysis. The safety set included all patients who received at least one administration of AUP1602-C. The evaluable set included all patients who received at least 75% of planned administrations during the first 4 weeks of treatment.
Demographics, diabetes history, and baseline ulcer characteristics (Safety set, n = 16).

Wound classification system according to Armstrong et al. 36
BMI, body mass index; HbA1c glycosylated hemoglobin; n, number of patients; SD, standard deviation.
At baseline, all patients presented Grade 1A wounds according to the University of Texas classification. The wound classification did not change in any patient after baseline debridement.
At baseline, the amount of exudation was assessed as moderate in two patients of Cohort 1 and in one patient of Cohort 3, and as low in the rest of the patients.
The mean (SD) 2D wound areas measured by the investigator at baseline were 2.3 (0.8), 6.3 (2.8), 1.9 (0.9), and 2.0 (0.8) cm2 for Cohort 1, Cohort 2, Cohort 3, and Cohort 4, respectively. The 3D wound areas assessed by an independent observer at baseline were in a similar range with mean (SD) of 3.0 (1.2), 7.2 (n = 1), 1.9 (1.1), and 2.1 (0.7) cm2 for Cohorts 1–4.
At baseline, none of the patients presented signs of local wound infection, systemic signs and symptoms of infection, or complications of infection. All target ulcers were assessed as non-ischemic ulcers and did not involve bone or joint.
Further data on wound assessment at baseline is given in Supplemental Table 2.
Extent of exposure
In Cohort 1, all three patients received the planned single IMP administration. In Cohort 2, patients received a mean (SD) of 24.7 (6.1) IMP administrations over a mean of 56.0 (14.1) days. Two patients extended their treatment period beyond 18 IMP administrations, 1 with 6 additional administrations, and 1 with 11 full and 1 partial (i.e., less than planned dose) additional administrations. In Cohort 3, patients received a mean (SD) of 19.3 (10.7) IMP administrations over a mean of 43.3 (24.8) days. Two patients extended their treatment period beyond 18 IMP administrations, 1 with 6 additional administrations and 1 with 12 additional administrations. One patient interrupted IMP due to a TEAE. In Cohort 4, patients received a mean (SD) of 16.5 (1.4) IMP administrations over a mean of 37.7 (3.5) days. None of the patients extended the treatment period. Two patients interrupted the IMP due to the wound being closed and three patients skipped a single IMP administration due to periwound skin maceration.
Safety endpoints
The primary objective of the study was to determine the safety and tolerability of AUP1602—C treatment. TEAEs reported during the study are summarized in Table 2.
Summary of TEAEs (Safety set, n = 16).
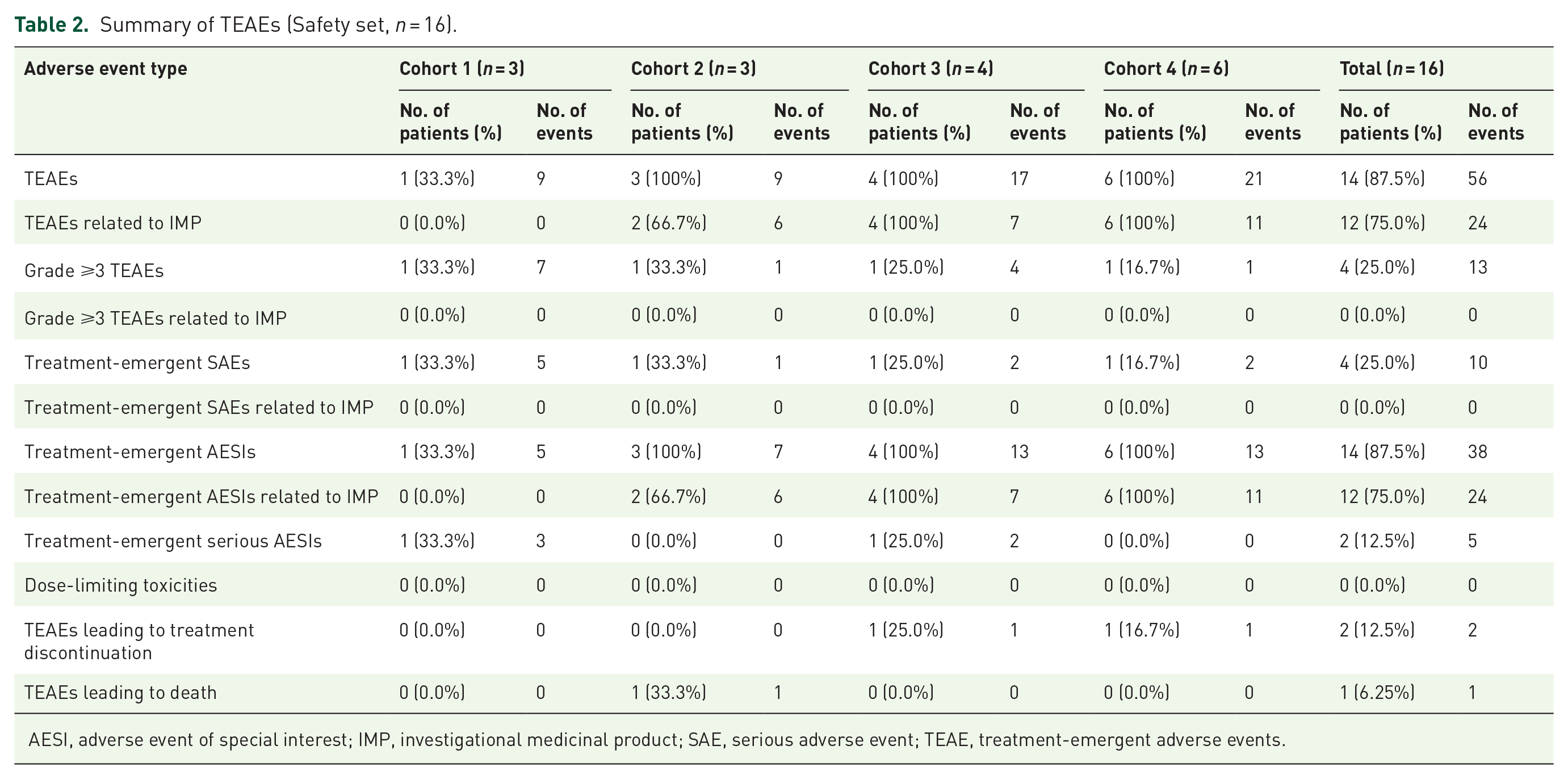
AESI, adverse event of special interest; IMP, investigational medicinal product; SAE, serious adverse event; TEAE, treatment-emergent adverse events.
Overall, 14 of 16 patients (87.5%) across all cohorts experienced a total of 56 TEAEs, 24 of which were assessed as related to the IMP occurring in 12 patients (75%). The most frequently reported TEAEs assessed as related to the IMP and as an adverse event of special interest (AESI) were skin maceration (22 events in a total of 12 patients), which is an expected AE based on risk–benefit assessment, due to the use of film dressings as the primary wound dressing. However, most skin maceration events (n = 20) were mild (NCI-CTCAE Grade 1) and resolved within a few days. Other IMP-related TEAEs were wound odor events recorded as wound complication (1 patient in Cohort 4) and hematoma (1 patient in Cohort 4) after wound biopsy, see Table 3.
TEAEs by SoC and PT (Safety set, n = 16).

TEAEs assessed as IMP-related are given in bold and italics. AE of a certain PT is only counted once for each patient.
IMP, investigational medicinal product; PT, preferred term; SoC, system organ class; TEAE, treatment-emergent adverse events.
A total n = 10 serious adverse events (SAEs) were reported in four patients (25%): one patient in Cohort 1 experienced anemia, soft tissue inflammation, cellulitis, peripheral ischemia, and fistula. One patient in Cohort 2 died of a Coronavirus infection during the follow-up period. One patient in Cohort 3 developed soft tissue necrosis and peripheral ischemia, and for one patient in Cohort 4, soft tissue inflammation and ischemic stroke were reported. None of the SAEs was assessed as related to the IMP.
None of the TEAEs was categorized as Grade 4, while there were 12 TEAEs categorized as Grade 3, for one patient of Cohort 1 (two cases of anemia and cellulitis, one case each of soft tissue inflammation, fistula, peripheral ischemia), one patient of Cohort 3 (inflammation of wound, soft tissue necrosis, two cases of peripheral ischemia), and one patient of Cohort 4 (soft tissue inflammation). None of the Grade 3 TEAEs was assessed as related to the IMP.
With the maximum feasible dose concentration applied, no DLTs were reported and therefore, no MTD was established and defined in this study. Two patients (12.5%) discontinued study treatment prematurely because of TEAEs (soft tissue inflammation, inflammation of the wound) which were assessed as not related to the IMP.
A total of 38 AESIs were reported in 14 patients (87.5%), of which 24 were assessed as related to study treatment. Five and 7 AESIs were reported in Cohorts 1 and 2, respectively, while 13 each were reported in Cohorts 3 and 4.
Results from laboratory assessments for hematology, biochemistry, coagulation, and urinalysis showed sporadic values outside the normal range. However, only two of these were reported as TEAE (anemia in one patient of Cohort 1). In general, no trend for an impact of the study medication on laboratory parameters was observed. There were no other safety signals noted, such as clinically significant abnormal values in vital signs, physical examinations, or other observations related to safety. Some patients showed abnormal echocardiogram findings at baseline which were assessed as not clinically significant, and no evidence of active infective endocarditis was detected during the study. Further, as expected in this controlled diabetic population, no clinically significant abnormal findings attributable to the treatment were observed in ophthalmoscopic assessments of retinopathy across visits, including at baseline.
There was no evidence of local wound infection at any patient visit, except in one patient of Cohort 3 who was treated with antibiotics. Overall, two TEAEs termed “inflammation around target ulcer” and one TEAE termed “cellulitis” were reported in three patients. One event of inflammation and one event of cellulitis associated with antibiotic therapy were reported in relation to the target ulcer. Both events were assessed as not related to treatment with AUP1602-C. One event of inflammation assessed as unlikely to be related to IMP treatment and one event of wound odor assessed to be possibly related to AUP1602-C treatment were also reported. Both these events were not associated with wound infection or antibiotic therapy. No local surgical procedures with the indication target ulcer were reported in any of the patients throughout the study, except for one patient who had developed wound inflammation around the target ulcer. The IMP was interrupted in this patient, but the condition progressed to necrosis and peripheral ischemia, and ended up in toe amputation.
Efficacy endpoints
By the end of treatment, 1/3 of patients (33.3%) in Cohort 3 and 3/5 patients (60%) in Cohort 4 had achieved complete wound closure. Additionally, at 10 weeks after the start of treatment, 1/3 of patients (33.3%) in Cohort 2 were reported with complete wound closure. At 6 months, complete wound closure was observed in 1/3 patients (33.3%) of Cohorts 1, 2, and 3 and in 5/6 patients (83.3%) of Cohort 4 (see Table 4). The median time to complete wound closure was 65 days in Cohort 4 and could not be estimated in Cohort 1–3 (Figure 3(b)). None of the patients who achieved complete wound closure experienced ulcer recurrence up to 12 months after the last IMP administration.
Summary of efficacy data (Evaluable set, n = 15).

Wound volume reduction >100% indicates hypergranulation.
FU, follow-up; SD, standard deviation.

Wound area reduction and time to complete wound closure. (a) Wound area changes during the 2-week run-in period under SoC (Screening to Visit 1) and during the first 2 weeks of treatment with AUP1602-C (Visit 1 to Visit 6). Data are presented as mean ± SEM. (b) Percentage and time for each cohort to reach total wound closure. Horizontal dotted line represents the 50% cohort level, and vertical dotted lines represent the end of AUP1602-C treatment and extension treatment.
Across all cohorts, the target wound’s size was reduced following AUP1602-C administration. At 10 weeks after the first administration, the mean reduction of 2D wound area, as assessed by the investigator, was 14.0%, 45.3%, 53.1%, and 76.8% in Cohort 1–4, respectively. At the 6 months follow-up visit, the mean reduction was 8.6%, 53.0%, 49.7%, and 97.1% in Cohort 1–4, respectively (Table 4, Figure 4). Comparable results were observed in the 3D assessment performed independently. Overall, the mean WAR in all treatment groups after the first 2 weeks of treatment with the IMP was higher than in the run-in period in which only SoC had been provided (Figure 3(a)). Per patient wound pictures are provided in Supplemental Figure 1.

Wound area reduction as assessed by the investigator (% from first AUP1602-C administration at each timepoint) and indication if and on which study day an individual patient achieved total closure. For Cohort 1, the single AUP1602-C administration was performed at timepoint 0 (a), whereas the vertical dotted lines represent the end of AUP1602-C treatment at 6 or up to 10 weeks for Cohorts 2–4 (b–d).
Compliance with off-loading
The mean (SD) off-loading compliance between screening and the 3-months follow-up visit based on hours was 92.9% (2.7%), 92.0% (2.3%), 81.2% (4.9%), and 45.7% (20.8%) in Cohort 1, Cohort 2, Cohort 3, and Cohort 4, respectively. Overall, all patients in Cohorts 1, 2, and 3 were >75% compliant, while patients in Cohort 4 were 50%–75% compliant (n = 2) or <50% compliant (n = 4). Regarding off-loading compliance based on steps, only one patient in Cohort 4 had information available, whose compliance was 77.3%.
Between the 3-month and 12-month follow-up visits, the mean (SD) off-loading compliance based on hours was 94.3% (5.0%), 76.1% (22.7%), 43.0% (33.4%), and 41.1% (45.5%) in Cohort 1, Cohort 2, Cohort 3, and Cohort 4, respectively, with 100%, 66.7%, 33.3%, and 33.3% of patients showing >75% compliance in the different cohorts.
Wound exudation
On average, the exudation status was assessed to be low in more than 70% of treatment visits in all repeat-dose study cohorts. Moderate wound exudation was observed between 15% and 25% of treatment visits, while high wound exudation was observed in less than 5% of treatment visits. 6/13 patients who received repeated AUP1602-C treatment experienced an increase in the amount of wound exudation from the last pretreatment assessment to the first post-treatment assessment. Overall, no dose relationship in the increase of wound exudation levels was observed with increasing AUP1602-C doses.
Quality of life
Results from EQ-5D-5L and DLQI questionnaires revealed a clear improvement in Cohort 4 as compared to Cohort 1, while Cohort 2 and 3 showed intermediate values. In Cohort 1, EQ-5D-5L mean scores by category (Mobility, Self-care, Usual activity, Pain/discomfort, Anxiety/depression) did not change over time, while in Cohort 4 score decrease was seen in all categories, particularly in mobility from 3.2 at S-V1 to 1.8 at FU-V6, pain, and discomfort from 2.8 at S-V1 to 1.0 at FU-V6 (Figure 5). The mean (SD) EQ-5D-5L utility index score (data now shown) increased by 0.1 from baseline to FU-V6 (8 weeks post last dose) to reach 1.0 (0.0) (best health state) in Cohort 4 versus decreasing by 0.1–0.8 (0.2) in Cohort 1. Mean EQ-5D-5L VAS increased by 10.0 (26.5) from baseline in Cohort 4 versus increasing by 1.7 (7.6) in Cohort 1. Mean DLQI score decreased by 5.6 (4.2) from baseline in Cohort 4 versus 1.7 (1.2) in Cohort 1 (Figure 6). Mean pain intensity decreased by 3.8 (2.4) from baseline in Cohort 4 versus increasing by 1.3 (1.5) in Cohort 1 (Figure 7). These results suggest a positive impact of AUP1602-C on QoL and pain perception for patients suffering from DFU.
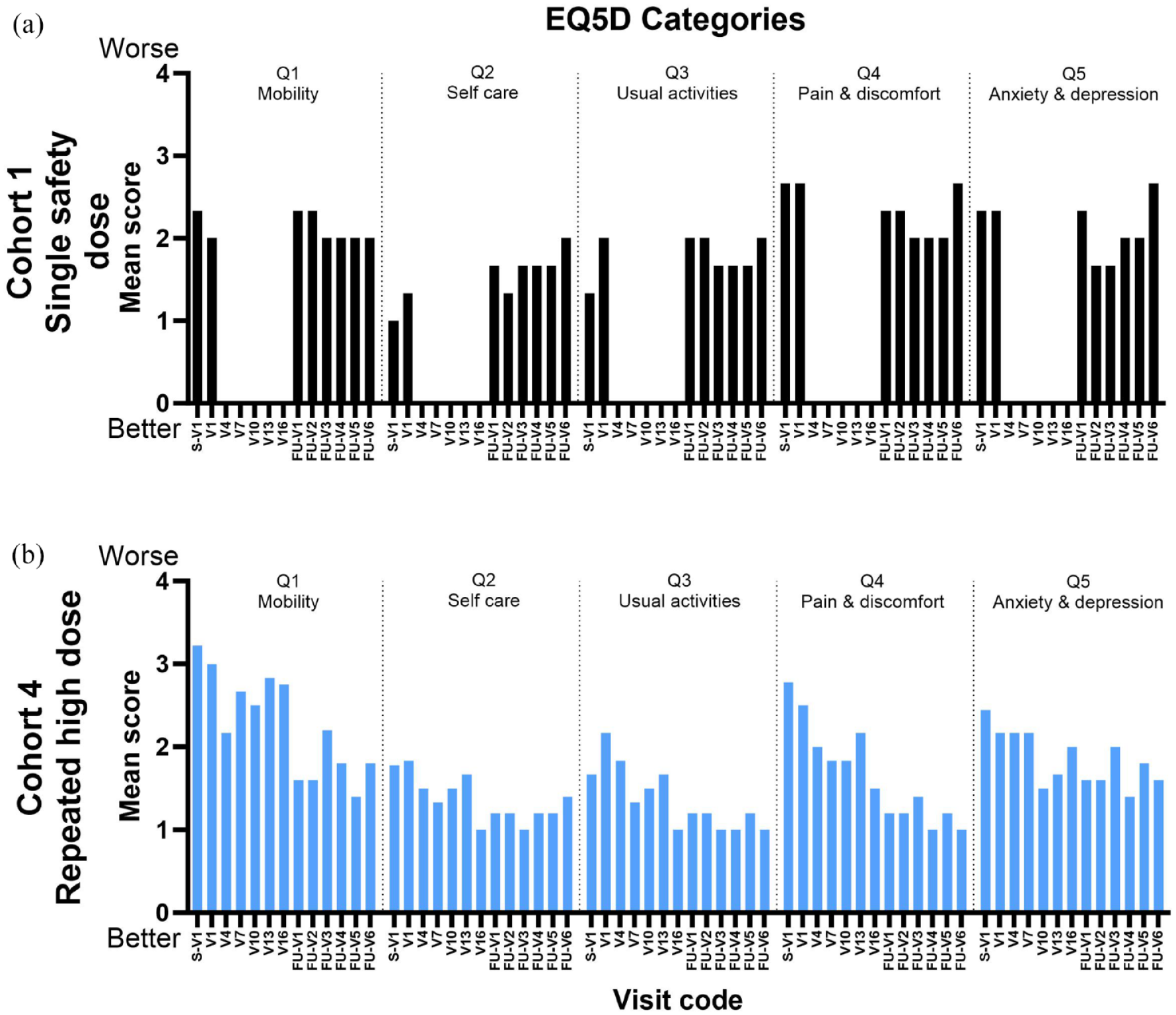
The five dimensions of EQ-5D-5L standardized measures of health-related quality of life. Data are presented as mean scores from worse (4) to better (0) at each time point for Cohort 1 (a) and Cohort 4 (b).

The DLQI measured at each time point with calculated MCID. The horizontal dotted line represents the MCID level which was calculated using the mean values of Cohort 1 score—5 points. Vertical dotted lines represent the treatment period. Data is presented as mean.

The pain perception score as measured by questionnaire at each time point. Vertical dotted lines represent the treatment period. Data is presented as mean.
Biodistribution and shedding
With 87.5% of planned biodistribution and shedding samples collected and analyzed, 93% (355 samples) were found to be negative, and 7% of analyzed samples (28 samples) were positive for AUP1602-C (plasmid). All positive events were shedding events and no biodistribution in plasma was observed. The 28 positive shedding events occurred in 6 urine samples and in 22 feces samples across all cohorts. In the positive samples, the amount of AUP1602-C (plasmid) was low and was close to the limit of detection of the assay (1 CFU).
Overall, across all positive samples tested, no apparent pattern for accumulation or dose dependency (i.e., no significantly increased number of positive events or significantly higher amount of AUP1602-C plasmid was observed between cohorts, even though different patient samples were treated with a dose escalation measured on a logarithmic scale) of AUP1602-C was observed. No systemic absorption was detected in the blood samples independently of the wound size across all patients tested. The number of positive events and the CFU of analyzed samples do not indicate any correlation with dosing and treatment frequency.
Discussion
Unmet medical needs resulting from limitations of SoC necessitate the development of advanced/adjuvant wound care products. Out of many advanced wound healing products for DFU, so far only growth factors and tissue-based substitution components have gained approval in the United States and other major countries. These include recombinant human platelet-derived growth factor Regranex® (Ortho-McNeil-Janssen Pharmaceuticals, New Jersey, USA) 37 ; three recombinant human epidermal growth factors: Easyef® (Daewong Pharmaceuticals, Seoul, South Korea), 38 Heberprot-P® (Heber Biotec, Havana, Cuba), 39 and Regen-D™150 (Bharat Biotech, Hyderabad, India) 40 ; the bilayered living skin construct Apligraft® (Organogenesis Inc., San Diego, USA) 41 ; the human fibroblast-derived dermal substitute Dermagraft® (Organogenesis Inc., San Diego, USA) 42 ; and the bilayer dermal regeneration matrix Omnigraft® (Integra Lifesciences, New Jersey, USA). 43 Among them, only Regranex (becaplermin) achieved EU approval in 1999 but was voluntarily withdrawn in 2012 by the applicant for commercial reasons.
The multitargeting gene therapy technology AUP1602-C, tested in this trial, allows the delivery of multiple therapeutic proteins that can engage different wound healing targets, such as angiogenesis, fibroblast proliferation, and modulation of the wound’s immune microenvironment.
This FIH, uncontrolled, open-label study employed a classical 3 + 3 dose escalation design to explore the MTD and DLTs, and to identify the recommended phase II dose of AUP1602-C. The principle of dose selection was based on the number of bacteria required to produce sufficient therapeutic proteins to demonstrate efficacy in a unit wound area, irrespective of the species. Based on this principle, the number of bacteria required per square centimeter of wound area is equal across different species including humans. The AUP1602-C starting dose of 2.5 × 105 CFU/cm2 ulcer size was selected based on non-clinical safety and toxicology studies ensuring a large safety margin for FIH used in the defined patient population. In addition, a scattered treatment schedule was applied, where the treatment for the second and third patient of a cohort was started 1 week after starting treatment and observation for DLTs of the first patient to further ensure patient safety. Three times a week as treatment frequency was selected for the study to provide maximum IMP exposure practically possible in a clinical setting. Treatment duration of 6 weeks was considered sufficient to evaluate DLTs. However, the investigators were allowed to extend the treatment duration to a maximum of 10 weeks if there was clinical evidence of benefit for the patient. A long-term safety follow-up period of 12 months after the last IMP treatment ensured monitoring of patient’s safety as per GTMP requirements and to evaluate the durability of the healing process. Complete wound closure and time to complete wound closure were selected as main efficacy parameters per regulatory guidelines. 44
During the FIH study, no DLTs were observed within the investigated dose range, and the MTD was not reached. Further dose escalation was not feasible due to technical limitations in producing AUP1602-C drug products in higher concentrations. Meanwhile, the highest dose tested, 2.5 × 108 CFU/cm2, demonstrated early signs of clinical efficacy. Therefore, this dose was selected as the recommended phase II dose.
Safety results were evaluated based on all patients who received at least one dose of AUP1602-C. AUP1602-C was very well tolerated by all patients. No safety issues related to the applied doses, duration of therapy, and time course of treatment with AUP1602-C were identified. No suspected unexpected serious adverse reactions, serious adverse reactions, evidence of systemic toxicity, TEAEs suggestive of immunogenicity, hypersensitivity, allergic reaction, DLTs, or deaths related to the treatment with AUP1602-C were reported.
Overall, no clinically relevant safety findings were identified during the study. The safety profile of AUP1602-C was consistent with the patients’ diagnosis of type 2 DM, and the risks were largely predictable, monitorable, and treatable in an appropriately equipped medical environment. Periwound skin maceration, the most common TEAE encountered during the study, is expected to be reduced by reducing the dosing frequency and contact time with film dressing. This approach is being further investigated in the ongoing phase II study in Europe.
Efficacy results were evaluated based on patients receiving a sufficient number of AUP1602-C applications to be eligible for DLT evaluation. Based on wound closure assessments, early signs of clinical benefit of AUP1602-C could be demonstrated. By the end of treatment, one patient (33.3%) in Cohort 3 and three patients (60%) in Cohort 4 had achieved complete wound closure. In addition, one patient (33.3%) in Cohort 2 showed complete healing at 10 weeks after the first treatment. At the last efficacy follow-up, that is, 6 months after end of treatment, 83% of patients in the high-dose cohort had achieved complete healing within a median time of 65 days. Three other patients, one in the safety, low, and medium dose cohorts, respectively, achieved total wound closure during the study. Overall, total wound closure in all patients receiving AUP1602-C was 53%, whereas 58% of patients receiving repeated AUP1602-C treatment achieved complete wound closure. Of note, none of the patients with complete wound closure experienced ulcer recurrence up to 12 months after the last IMP administration. The follow-up time was chosen to ensure that key efficacy endpoints could be presented in accordance with regulatory recommendations. 45
A post hoc comparison of WAR during the first 2-weeks of AUP1602-C treatment against the 2-weeks of SoC treatment during the run-in period revealed a 30% average WAR with AUP1602-C treatment, as opposed to a 17% increase with SoC within the same period. Across all cohorts, target wound size had decreased after AUP1602-C administration, with a mean reduction ranging from 14.0% in Cohort 1 (single safety dose) to 76.8% in Cohort 4 (multiple, high dose) at 10 weeks after start of treatment. Similar results were observed regarding wound depth and wound volume, indicating a dose–response relationship that should be further investigated in larger randomized controlled studies. Overall, data from this FIH study supports selection of 2.5 × 108 CFU/cm2 as recommended phase II dose.
The reduction in off-loading compliance observed in Cohort 4 compared to the other cohorts could possibly be attributed to the early complete wound closure observed in this cohort. However, it has to be noted that self-reported off-loading compliance is often overestimated. 46
Data on health-related QoL and pain perception revealed clear improvement at the recommended phase II dose as compared to the single dose, showing evidence for a beneficial effect of AUP1602-C in patients suffering from DFU.
The limitations of this FIH study were the uncontrolled, non-randomized, open-label study design. Furthermore, the study was conducted on a small number of patients at a single study site, which limits generalizability of the study results. However, a Phase 2 study including a larger patient population is ongoing in Europe, which will provide further data for external validation of the study results reported here.
Conclusion
To our knowledge, this was the first clinical study using a bacteria-based, multi-target gene therapy that accelerated wound healing and demonstrated clinical benefit in patients with non-healing or hard-to-heal neuro-ischemic DFU. Overall, AUP1602-C showed a favorable safety profile, evidence for efficacy, and a positive impact on QoL. Data available to date supports further clinical development of AUP1602-C with a recommended phase II dose of 2.5 × 108 CFU/cm2.
Supplemental Material
sj-doc-1-tae-10.1177_20420188241294134 – Supplemental material for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study
Supplemental material, sj-doc-1-tae-10.1177_20420188241294134 for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study by Christoph Schindler, Jacek Mikosiński, Pawel Mikosiński, Hanna-Riikka Kärkkäinen, Mirka Sanio, Jere Kurkipuro, Igor Mierau, Wesley Smith, Aki Vartiainen, Laurent Décory, Dirk Weber, Thomas Wirth, Juha Yrjänheikki, Sebastian Schellong and Haritha Samaranayake in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-docx-2-tae-10.1177_20420188241294134 – Supplemental material for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study
Supplemental material, sj-docx-2-tae-10.1177_20420188241294134 for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study by Christoph Schindler, Jacek Mikosiński, Pawel Mikosiński, Hanna-Riikka Kärkkäinen, Mirka Sanio, Jere Kurkipuro, Igor Mierau, Wesley Smith, Aki Vartiainen, Laurent Décory, Dirk Weber, Thomas Wirth, Juha Yrjänheikki, Sebastian Schellong and Haritha Samaranayake in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-docx-3-tae-10.1177_20420188241294134 – Supplemental material for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study
Supplemental material, sj-docx-3-tae-10.1177_20420188241294134 for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study by Christoph Schindler, Jacek Mikosiński, Pawel Mikosiński, Hanna-Riikka Kärkkäinen, Mirka Sanio, Jere Kurkipuro, Igor Mierau, Wesley Smith, Aki Vartiainen, Laurent Décory, Dirk Weber, Thomas Wirth, Juha Yrjänheikki, Sebastian Schellong and Haritha Samaranayake in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-docx-4-tae-10.1177_20420188241294134 – Supplemental material for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study
Supplemental material, sj-docx-4-tae-10.1177_20420188241294134 for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study by Christoph Schindler, Jacek Mikosiński, Pawel Mikosiński, Hanna-Riikka Kärkkäinen, Mirka Sanio, Jere Kurkipuro, Igor Mierau, Wesley Smith, Aki Vartiainen, Laurent Décory, Dirk Weber, Thomas Wirth, Juha Yrjänheikki, Sebastian Schellong and Haritha Samaranayake in Therapeutic Advances in Endocrinology and Metabolism
Supplemental Material
sj-pptx-5-tae-10.1177_20420188241294134 – Supplemental material for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study
Supplemental material, sj-pptx-5-tae-10.1177_20420188241294134 for Multi-target gene therapy AUP1602-C to improve healing and quality of life for diabetic foot ulcer patients: a phase I, open-label, dose-finding study by Christoph Schindler, Jacek Mikosiński, Pawel Mikosiński, Hanna-Riikka Kärkkäinen, Mirka Sanio, Jere Kurkipuro, Igor Mierau, Wesley Smith, Aki Vartiainen, Laurent Décory, Dirk Weber, Thomas Wirth, Juha Yrjänheikki, Sebastian Schellong and Haritha Samaranayake in Therapeutic Advances in Endocrinology and Metabolism
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.