
Abstract
Metabolic dysfunction–associated fatty liver disease (MAFLD) prevalence and incidence is rising globally. It is associated with metabolic comorbidities, obesity, overweight, type 2 diabetes mellitus, and at least two metabolic risk factors, such as hypertension, hypertriglyceridemia, hypercholesterolemia, insulin resistance, and cardiovascular risk, increasing the risk of mortality. The excessive accumulation of fat comprises apoptosis, necrosis, inflammation and ballooning degeneration progressing to fibrosis, cirrhosis, and liver decompensations including hepatocellular carcinoma development. The limitation of approved drugs to prevent MAFLD progression is a paradigm. This review focuses on recent pathways and targets with evidence results in phase II/III clinical trials.
Introduction
Nonalcoholic fatty liver disease (NAFLD) has become a serious public health problem with a prevalence of 25% 1 and is a leading cause of cirrhosis, liver transplantation, and hepatocellular carcinoma.2,3 NAFLD is a multifactorial, complex disease associated with metabolic and cardiovascular disorders, obesity, type 2 diabetes (T2DM), insulin resistance (IR), hypertension, and dyslipidemia. In fact, NAFLD is recognized as the hepatic manifestation of the metabolic syndrome.4,5 In recent times, a new definition was suggested for NAFLD, namely metabolic dysfunction–associated fatty liver disease (MAFLD), in which there are several criteria for the diagnosis: hepatic steatosis detected by image test, liver histology or noninvasive biochemical tests, and without any association with significant alcohol intake, defined as 30 g/day for women and 40 g/day for men; long-term use of steatogenic medication; or monogenic hereditary disorders, associated with metabolic disorders, overweight, obesity, and T2DM. 6
MAFLD spectrum ranges from simple steatosis, characterized by the accumulation of lipids in more than 5% of hepatocytes, to the more aggressive phenotype, nonalcoholic steatohepatitis (NASH), histologically characterized by the presence of steatosis, hepatocyte injury (ballooning), and inflammation with or without fibrosis. 7 The pathogenesis of metabolic liver diseases involves lifestyle (nutritional overload and physical activity), and genetic and environmental factors.
The gold standard for MAFLD diagnosis is liver biopsy, an invasive method that carries risk and is expensive. Nowadays, noninvasive scoring tools are being developed for NAFLD patient stratification such as NAFLD fibrosis score. Whereas, liver biopsy histology results allow to create the NAFLD Activity score (NAS Score) or Steatosis Activity Fibrosis (SAF) score. Both quantify steatosis, hepatocyte ballooning, and lobular inflammation. 8 In addition, NASH Clinical research network score (NASH CRN) includes the fibrosis stage, and both are very useful to validate the efficacy of treatments.9,10
There is no approved drug for NASH to date, so current treatment consists on the reduction of body weight through lifestyle interventions.11,12 New drug development focuses on the restitution of metabolic derangements and halting inflammatory and fibrogenic pathways. This review summarizes different pathways under study for MAFLD treatment and the emerging therapies already in phase II/III registration trials.
Crosstalk of multiple pathways in MAFLD
Metabolic pathways (lipid, glucose, and thyroid)
Steatosis is the consequence of imbalanced transportation, synthesis, and catabolism of fatty acids (FA) in the hepatocytes. 13 There is an increase in FA uptake and synthesis by hepatocytes and a reduction of FA mitochondrial β-oxidation together with decreased very low-density lipoprotein (LDL) secretion.14,15 The main sources of free fatty acids (FFAs) in the liver are diet, adipose tissue (lipolysis), and de novo lipogenesis (DNL). In NASH, DNL is increased being one of the most sources of fat. 16 Lipotoxicity caused by high lipid concentration in the hepatocytes results in insulin resistance, liver inflammation, cell injury, apoptosis, and fibrotic remodeling by the activation of hepatic stellate cells (HSCs).17,18
The hallmark of MAFLD is the accumulation of fat in the hepatocytes as lipid droplets (LDs). TGs are the major lipid class present in the LDs, but currently, they have been considered protective regarding cell toxicity. Diacylglycerol acyltransferase 1 and 2 (DGAT-2), the main enzyme responsible for TG synthesis, when inhibited results in a reduction of steatosis but with an increase in inflammation, oxidative stress, and fibrosis. 19 Different studies revealed that saturated FFAs were more toxic than unsaturated FFAs. In vitro studies demonstrated that palmitic acid (C16:0), the most common saturated FA, increases the number of LDs in the hepatocytes, activates the peroxisome proliferator-activated receptor alpha (PPAR-alpha), promotes insulin resistance, the stress of ER, and induces apoptosis, simulating the scenario of NASH.16,20 In NASH, there is an upregulation of HMG-CoA reductase, resulting in its accumulation mainly in the mitochondria and enhanced mitochondrial dysfunction, with an increase of ROS, leading to ER stress, apoptosis, and inflammation.21,22
A correlation of Bile Acids (BAs) levels with NASH severity has been described. 23 The farnesoid X receptor (FXR) negatively regulates BA synthesis and plays a crucial role in TGs, cholesterol, and glucose metabolism in the liver.24,25 FXR activation reduces lipotoxicity through inactivating the DNL and increases β-oxidation and cholesterol excretion, thus resulting in reduced IR, inflammation, and fibrosis. 26 FXR is a nuclear receptor as PPARs.25,27 PPARs (alpha, beta, and gamma) are expressed in the liver and peripheral tissues regulating multiple metabolic pathways such as β-oxidation, gluconeogenesis, and lipid transport. 27
Moreover, direct agents targeting molecular pathways of cholesterol and TGs synthesis could be considered good candidates as targets for MAFLD treatment. Acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), stearoyl-CoA desaturase 1 (SCD1), and DGAT catalyze the limiting step in DNL. ACC catalyses the carboxylation of acetyl-CoA to manolyl-CoA. 28 SCD1 catalyses the formation of monounsaturated fatty acids from saturated fatty acids 29 and unsaturated FA are esterified to produce TGs by DGAT1/2. 30
Hyperglycaemia induces hepatic fat accumulation contributing to ER stress signaling, progression of liver damage from simple steatosis to NASH and cirrhosis. 31 Glucagon-like peptide 1 receptor (GLP-1R) regulates blood sugar levels. GLP-1 treatment improves glycemic control, reducing body weight and IR, 32 being studied as MAFLD treatment.
MAFLD is also associated with thyroid hormone receptor beta (THRβ). THRβ is expressed in the human liver, and it is involved in lipid metabolism upregulating FFA uptake and oxidation: 33 THRβ induces BA synthesis and interacts with PPARs. 34 In fact, there are selective THRβ agonists that are being developed for MAFLD treatment.
Fibrotic pathways
The management and regression of liver fibrosis in MAFLD patients are one of the clinical endpoints of new drugs. Advanced fibrosis is the most significant predictor of mortality and liver cancer development in MAFLD. Improvement by at least one stage of fibrosis score is essential for the efficacy of drugs. HSCs are the principal cells responsible for collagen deposition in the liver. HSC activation is controlled by several signals such as lipotoxicity and inflammation, two scenarios very frequent in MAFLD. The development of anti-fibrotic drugs for MAFLD is one of the challenges in this area. Briefly, we summarize some targets related to fibrosis pathways.
FXR, also known as bile acid receptor, is also related to fibrogenesis. It is expressed in hepatocytes and Kupffer cells modulating HSC activation. 35 FXR exerts multiple beneficial metabolic effects, contributes to glucose regulation at the hepatic and peripheral level, is implicated in DNL and in fatty acid oxidation, and also exerts anti-inflammatory effects, 25 thereby influences hepatic metabolism, inflammation, and liver fibrosis, all of them, histological features of NASH. 36 FXR agonists improve the histological features of NASH and protect again liver fibrosis development in several animal models.37,38 FXR activation also improves vascular inflammation, remodeling, and sinusoidal vasodilation, improving portal hypertension in experimental models.39–41
Fibroblast growth factor families are constituted by FGF21, FGF19, FGF15, and FGF23. Cellular origin, expression, and regulation of FGF19 and FGF21 are not well understood. Fibroblast growth factor 21 (FGF21) is known to be highly expressed in the liver, and it is involved in liver glucose and lipid metabolism. 42 Altered FGF21 signaling is implicated in MAFLD pathology. FGF21 serum levels were increased in NASH patients. 43 In contrast, lower serum levels of FGF19 were reported in biopsy-proven MAFLD patients independently of liver damage severity, 44 and levels of FGF19 were inversely associated with disease severity. 45
Ballooned hepatocytes are a hallmark of NASH and fibrosis progression and represent the activation of apoptosis pathways. Apoptosis signal-regulating kinase (ASK) 1 is implicated in the response to oxidative and ER stress. 46 ASK1 inhibition prevents liver inflammation, fibrosis, and cell death. 47 ASK1 deficiency protects against liver damage caused by acetaminophen or under stress conditions such as high fat diet, thus highlighting the therapeutic potential.48,49
Lysyl oxidase-like 2 (LOXL2) plays an essential role in matrix remodeling and fibrogenesis. LOXL2 promotes covalent cross-linking of elastin and collagen fibers, indicating an essential role in fibrosis-associated liver disease and limits its resolution. 50 LOXL2 was absent in healthy but strongly expressed in fibrotic liver (predominantly in fibrotic septa) in a chronic thioacetamide (TAA) administration animal model. 51 Furthermore, previous studies showed an improvement in liver fibrosis in a mouse model of mild fibrosis after early treatment with anti-LOXL2 antibody. 52 Furthermore, delayed anti-LOXL2 treatment in mice significantly reduced collagen deposition and histological signs of fibrosis, promoting a reduction of advanced parenchymal liver fibrosis. Therefore, selective targeting of LOXL2 inhibits liver fibrosis progression and accelerates its reversal. 51
Inflammatory pathways
Fat hepatocyte injury increases several mechanisms of hepatic inflammation driving the progression of MAFLD and thus are under-studied as new targets for MAFLD. 53
C-C chemokine receptor type 5 (CCR5) organizes the hepatic recruitment, migration, and/or activation of immune cells as well as HSCs, with subsequent inflammation and fibrosis in MAFLD. 54 In mouse models of obesity, steatohepatitis, or liver fibrosis, the CCR2 has been unequivocally linked to the aggravation of inflammation and fibrogenesis.55,56 In addition, an increased number of cells expressing CCR2 has been observed in patients with chronic liver diseases and fibrosis. Furthermore, elevated numbers of CCR2+ macrophages are found in adipose tissue in patients with NASH.57,58
Monocyte recruitment into NASH liver can be effectively inhibited by the chemokine receptor CCR2/CCR5 inhibitor cenicriviroc (CVC). 59 CVC was associated with a higher rate of fibrosis improvement after 1 year of therapy.59,60 In animal models, CVC treatment improved IR and hepatic TGs levels and reduced histological NASH activity and hepatic fibrosis.
Galectin-3 plays a key role in apoptosis, adhesion, and immune response, and it has been implicated in the disease severity of NASH. 61 Preclinical models and clinical trials showed that targeting Gal-3 could reduce hepatic fibrosis. 62 Galectin inhibitors are a new class of agents that have been tested for MAFLD progression, such as belapectin which showed efficacy in preclinical models of NASH and liver fibrosis. 63
Emerging therapeutics of NASH
Numerous drugs with different targets have been developed in the past 15 years for MAFLD. Many of them are currently in development or preclinical studies or have already failed to show improvement in NASH features.
The goal of the emerging drugs is the reduction of fatty acid accumulation, reduction of inflammation and regression of fibrosis. The Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidance describe NASH resolution as the presence of any grade of steatosis, no ballooning, and only minimal (grade 1) lobular inflammation and – at the same time – no worsening of the stage of fibrosis; or the improvement of fibrosis by at least 1 stage without any worsening of NASH (no worsening of ballooning and lobular inflammation, a 1 grade change in steatosis may be acceptable).
In this review, we navigate through the emerging therapies advances in classes of drugs that are already in phase II/III clinical trials (Tables 1–5).
Clinical trial assessed lipid metabolism pathway.

ACC, acetyl-CoA carboxylase; DGAT2, diacylglycerol acyltransferase 1 and 2; FASN, fatty acid synthase; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NCT, National Clinical Trial.
PPARs agonist clinical trials.
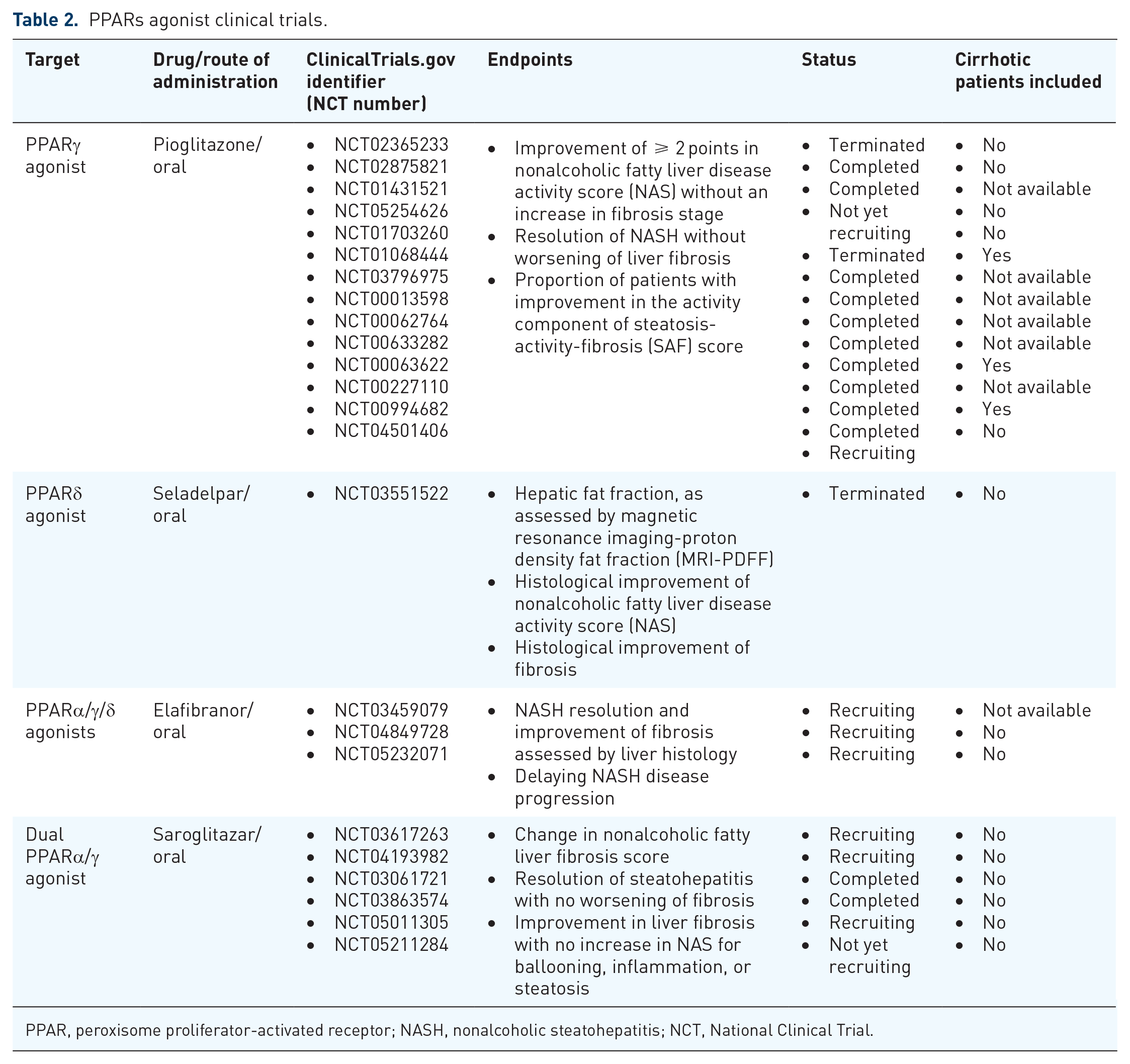
PPAR, peroxisome proliferator-activated receptor; NASH, nonalcoholic steatohepatitis; NCT, National Clinical Trial.
Clinical trials using incretins and thyromimetics drugs.

CRN, clinical research network; GCG, glucagon; GLP-1R, glucagon-like peptide 1 receptor; GIPR, glucose-dependent insulinotropic polypeptide receptor; LDL-C, low-density lipoprotein cholesterol; MRI-PDFF, magnetic resonance imaging-proton density fat fraction; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; NCT, National Clinical Trial; PD, proton density.
FXR agonists and FGF analogues trials.

ALT, alanine transaminase; AST, aspartate transaminase; CRN, clinical research network; FGF, fibroblast growth factor; FXR, farnesoid X receptor; LDL, low-density lipoprotein; NASH, nonalcoholic steatohepatitis; NCT, National Clinical Trial.
Antifibrotic drugs clinical trials.
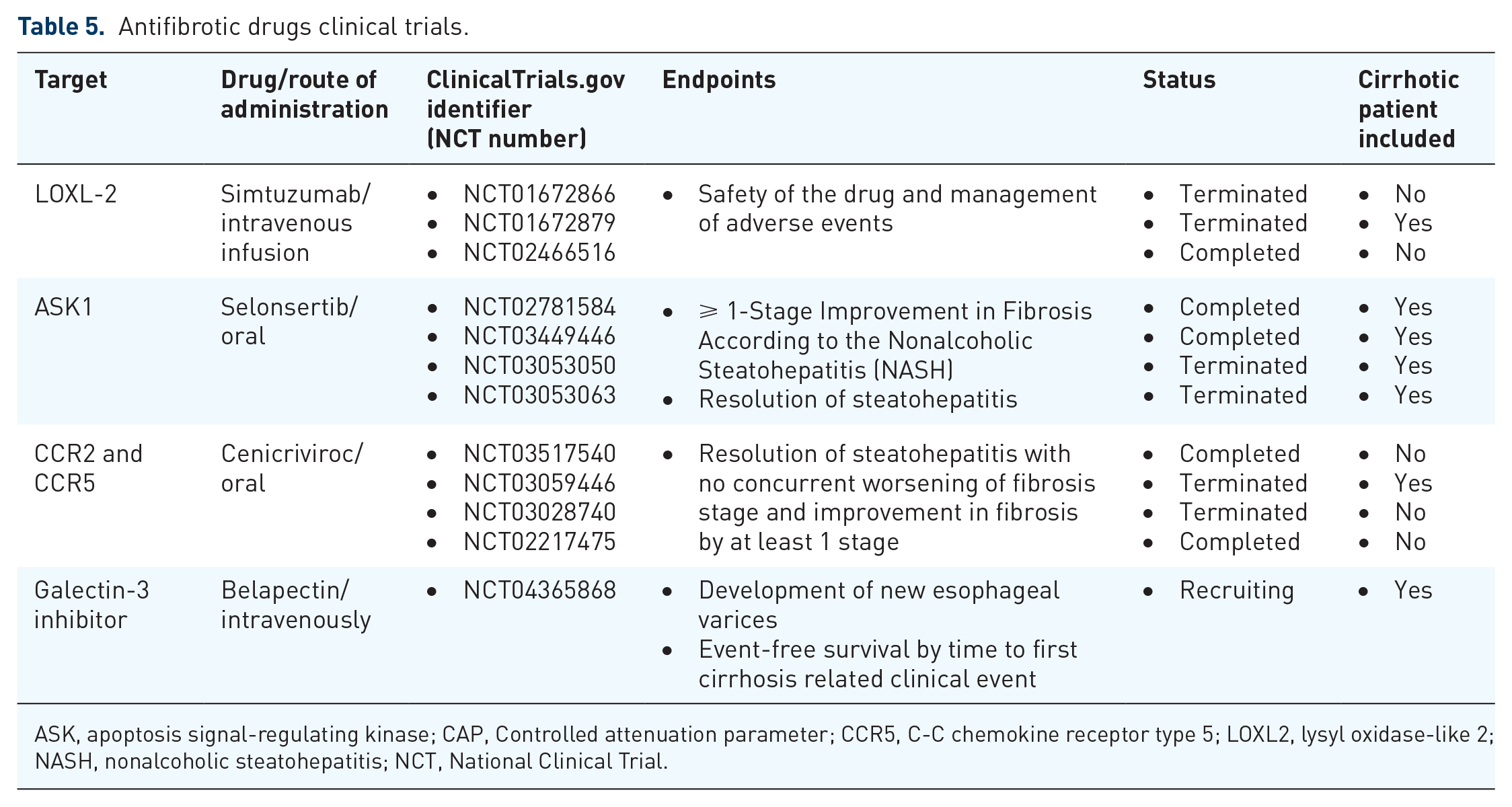
ASK, apoptosis signal-regulating kinase; CAP, Controlled attenuation parameter; CCR5, C-C chemokine receptor type 5; LOXL2, lysyl oxidase-like 2; NASH, nonalcoholic steatohepatitis; NCT, National Clinical Trial.
Lipogenesis inhibitors
PF-05221304 and Firsocostat are selective ACC inhibitors. Both of them have been tested in MAFLD patients alone or in combination with DGAT2 inhibitors or FXR agonist to maximize the resulting effects. The primary endpoint was changing the liver fat percentage measured by magnetic resonance imaging (MRI). Reductions in liver fat were reached at dose-dependent manner alone or in combination with DGAT2 inhibitor, 64 reducing the adverse events (AE) of the ACC inhibitor. In phase IIb, the combination of FXR agonist and ACC inhibitor provided significant reductions in NAS scores, liver steatosis, lobular inflammation and ballooning and improved biochemistry profile in NASH patients. 65 In this study, patients with advanced fibrosis were included to explore the improvement in fibrosis. Results showed changes in hepatic collagen deposition and a decrease in NASH CRN fibrosis score (p = 0.04). This combination would offer the potential for fibrosis regression in NASH patients with advanced fibrosis.
TVB-2640 is an FASN inhibitor that reduced liver steatosis in obese subjects with MAFLD risk. 66 In phase II, TVB-2640 at lower doses improved liver biochemistry and lipid profile, and attenuated steatosis and fibrosis biomarkers after 12 weeks. 67 TVB-2640 decreased serum fibrosis markers such as PRO-C3, TIMP-1, and PIIINP at 12 weeks, demonstrating that FASN inhibition could have an impact on HSCs and fibrogenesis pathways. A subsequent phase II study has been initiated to know the impact on the resolution of NASH without worsening of liver fibrosis.
Ervogastat (PF-06865571) is a selective DGAT2 inhibitor that reduces the liver fat fraction in patients with mild MAFLD. 68 Two clinical trials are underway to assess the safety and efficacy of ervogastat alone and in combination with ACC inhibitors in NASH patients with and without liver fibrosis. The primary outcome is the resolution of NASH without worsening of fibrosis or improvement in fibrosis by ⩾1.
ω-3 Polyunsaturated fatty acids (ω-3 PUFA) are long-chain FA with a double bond three atoms away from the terminal methyl group. ω-3 PUFA includes α-linolenic acid and its metabolites eicosapentaenoic and docosahexaenoic acids. Recent meta-analyses included up to 22 randomized control trials (RCT) and more than 1300 patients and found that ω-3 PUFA significantly decreases liver transaminases, liver fat, and IR, having no effect on body weight in MAFLD.69–71 There are also artificial ω-3 PUFAs, icosabutate (NST-4016), being studied in a phase II trial in patients with biopsy-confirmed NASH. Interim analysis data indicated improvements in noninvasive fibrosis and inflammatory biomarkers. 72
PPAR agonists
Pioglitazone, a selective PPARγ agonist, is supported by the European Association for the Study of the Liver (EASL) and American Association for the Study of the Liver (AASLD)8,73 clinical practice guidelines, due to its efficacy in the liver histology in biopsy-proven NASH patients. Pioglitazone improves ballooning degeneration, lobular inflammation, steatosis, and fibrosis. 74 Although there are several phase IIb trials,75,76 there are no phase III trials to demonstrate pioglitazone’s histological efficacy. Furthermore, several AE such as fluid retention, weight gain, and bone loss have questioned its long-term use in NASH.
Seladelpar (MBX-8025) is the only selective PPARδ agonist currently in development for the treatment of MAFLD. The interim analysis results of a 52-week phase IIb RCT showed minimally influence on liver steatosis at 12 weeks of treatment. 77
Lanifibranor is an experimental triple PPARα/γ/δ agonists. Its predecessor, elafibranor (GFT-505), was discontinued due to lack of efficacy in MAFLD in the phase III. Lanifibranor was well tolerated, and the percentage of patients with meaningful improvements in steatosis, activity, and fibrosis scores was significantly higher in the lanifibranor treated arms in a completed phase IIb study with 247 patients. 78 Two more trials to evaluate the efficacy of lanifibranor in concomitant MAFLD and T2DM and advanced fibrosis due to NASH are ongoing.
Saroglitazar, a dual PPARα/γ agonist, had been tested in MAFLD patients with or without T2DM, and it has shown promising results in western trials with an improvement in liver biochemistry as well as hepatic steatosis.79,80 It is already approved in India for use in T2DM and pre-cirrhotic NASH. At 12 weeks of treatment, saroglitazar improved clinical parameters such as glucose, HbA1c, total cholesterol, TGs, and liver stiffness when compared with the baseline values. 81
Incretins
GLP-1 receptor agonists are indicated and accepted by the FDA for obesity and T2DM. Semaglutide is being tested for the treatment of NASH in nondiabetic subjects. In the recently completed 72-week phase II trial, semaglutide treatment achieved the highest response rate in NASH resolution in a trial until now without worsening of fibrosis.82,83 However, there was a lack of fibrosis reversal despite the massive weight loss so there is the question if the effects are weight loss-independent effects.
Liraglutide is another GLP1R agonist that has demonstrated a hepatitis activity reduction and fibrosis reduction in the phase II study. 84 However, the small sample size and the lower mean body mass index (BMI) in the placebo group were important limitations in this study. Nevertheless, improvement in liver fat content in patients with T2DM was also observed in the Lira-NAFLD study. 85 In a meta-analysis of eight clinical trials, it was shown that GLP1R agonist could improve histology in T2DM NAFLD patients and liver function with a reduction of BMI, liver fat concentration, and glycaemia levels. 86
In addition, dual agonists have been studied for NASH treatment, associating GLP-1 with GIP agonism (tirzepatide) 87 or GLP-1 with glucagon (GCG) agonism (cotadutide). 88 There are also novel triple GLP1R/GCGR/GIPR agonists being evaluated, such as HM15211. 89
Thyromimetics
Selective THRβ agonists that are currently being developed for the treatment of NAFLD include resmetirom and VK2809. Resmetirom is the first oral, liver-directed THR-β1-selective agonist. In a 36-week phase II randomized clinical trial, resmetirom achieved NASH resolution in a subset of patients with control biopsies. Liver steatosis and liver stiffness improved together with lipid serum profile and fibrosis biomarkers such as Pro-C3 and hepatic enzymes, whereas a significant reduction in NAFLD activity was observed. 90 VK2809 is another THRβ agonist which is metabolized in the liver by CYP450. It showed a very good tolerability profile, and a significant reduction in liver fat was observed by MRI after 12 weeks of treatment. 91
FXR agonists
Obeticholic acid (OCA) is a first-in-class FXR agonist approved by the FDA for noncirrhotic primary biliary cholangitis (PBC) treatment. In fact, it is near to be approved for liver fibrosis in NASH. OCA reduces significantly alanine transaminase (ALT) serum levels, improves NAS scores, and induces histological regression of fibrosis compared with placebo in nondiabetic pre-cirrhotic NASH patients. 92 In a phase III trial, 14.9% of NASH patients with F1-F3 fibrosis improved NASH without worsening fibrosis. 93
But OCA is not exempt from side effects: pruritus and an increase in LDL concentration have been reported. FDA was to delay conditional approval of OCA until more efficacy and safety data are available, mainly concerning the increase of LDL and its possible cardiovascular effect. Second-generation FXR agonists are in development to avoid side effects.
MET409, a second-generation FXR agonist, has better efficacy and less pruritus and LDL levels than OCA. 94 There is an active phase IIa clinical trial to evaluate MET409 alone or with SGLT2 inhibitor (empagliflozin), but there are no results yet.
Tropifexor and cilofexor are FXR agonists with a different structure than OCA and MET409. In a pilot study, 10 patients with NASH and fibrosis (F2-F3) who received 30 mg cilofexor a nonsteroidal FXR agonist formerly GS-9674, for 12 weeks, experienced decreased hepatic fat, liver stiffness, and improved liver biochemistry. 95 In a recent phase-2b study, patients with NASH treated with cilofexor for 24 weeks showed a reduction in hepatic steatosis, serum bile acids, and an improvement in liver enzymes levels, but no significant changes regarding liver stiffness measured by transient elastography were observed. 96 In addition, pruritus was reported as AE. Tropifexor in biopsy-proven NASH patients with F2-F3 fibrosis reduced ALT, gamma-glutamyltransferase (GGT) levels, body weight, and liver fat content, and attenuated liver fibrosis in patients with biopsy-confirmed NASH in the 48-week phase II. However, LDL cholesterol and pruritus were the main adverse events producing discontinuation of treatment.97,98 More clinical trials with FXR agonists are ongoing to evaluate the safety, tolerability, and their role in complication events of cirrhosis. The results of the study are expected to be announced soon.
FGF analogues
Aldafermin is an engineered FGF19 analogue studied in NASH patients with liver fibrosis stage 2 or 3. It was well tolerated but it did not achieve the primary endpoint: improvement fibrosis defined as a ⩾1 NASH CRN stage without worsening of NASH. FGF19 analogue decreased fibrosis stage in 42% of subjects without NASH worsening and an improved of NAS without fibrosis worsening in 63% of patients.99,100 New trials are ongoing to determine whether aldafermin improves liver fibrosis in NASH subject with compensated cirrhosis.
Efruxifermin is an FGF21 analogue that significantly attenuated liver steatosis in the 16-week phase IIa in T2DM patients. 101 Efruxifermin is now being evaluated in three more phase II RCTs but no clinical data are available yet.
ASK1 inhibitor
Selonsertib, inhibitor of ASK1, reduces steatosis, fibrosis, and inflammation in NASH.102,103 The last clinical trial phase III has shown that ASK1 inhibitor was not better than placebo arm in terms of fibrosis stiffness improvement. 104
Anti LOX2
Simtuzumab, inhibitor of LOXL-2, was designed for fibrosis treatment. After 96 weeks of treatment in primary sclerosing cholangitis, hepatic collagen changed but without significant results. 105 Changes in hepatic venous pressure gradient (HVPG) at 96 weeks were measured in NASH patients with compensated cirrhosis, without efficacy. Simtuzumab did not decrease HVPG, fibrosis stages, or liver-related events. 106
CCR2 y CCR5 inhibitor
Efficacy and safety of Cenicriviroc (CVC) was evaluated in NASH with F2-F3 fibrosis stages. The endpoint was the improvement of ⩾ 1-stage in liver fibrosis and no worsening of NASH. 107 In a clinical study with pair-liver biopsy (baseline and 1 and 2 year), around 25% of patients achieved more or equal 1 stage of fibrosis and improved liver fibrosis. 108
Galectin antagonist
Although the involvement of galectin in chronic liver disease remains controversial, it seems that its increased expression is linked to accelerated cirrhosis development and worsening of liver function. 109 Hence, modern galectin-targeting drug candidates are intended for use in advanced NASH complicated by liver fibrosis and/or cirrhosis. Belapectin is an inhibitor of galectin-3 that has been evaluated in cirrhotic NASH patients with portal hypertension. In a 52-week phase IIb study, belapectin did not change fibrosis or NAFLD activity, but a significant reduction of HVPG and esophageal varices development was observed. 110 A new phase II/III trial has been initiated to evaluate belapectin in patients with liver cirrhosis due to NASH and clinical signs of portal hypertension but without esophageal varices at baseline.
Conclusion
Despite recent advances in the pathophysiology of MAFLD and the development of several drugs that are already close to be approved for MAFLD treatment, new strategies combining multitarget drugs need to be studied.
Footnotes
Acknowledgements
Ministerio de Ciencia y Competitividad under grant agreements PI19/01404 and PI19/00589. A contract with fellowship from Consejería de Salud y Familias, Junta de Andalucía, supporting Angela Rojas (RH-002-2021); grant agreements PFIS FI20/00201 from Instituto de Salud Carlos III to support Sheila Gato and by support from Talento Doctores (PID Junta Andalucía, DOC_00866) to Rocío Muñoz-Hernández.