
Abstract
Polycystic ovary syndrome (PCOS) is the most common endocrine condition affecting women. It has traditionally been viewed as a primarily reproductive disorder; however, it is increasingly recognized as a lifelong metabolic disease. Women with PCOS are at increased risk of insulin resistance (IR), type 2 diabetes mellitus, non-alcoholic fatty liver disease and cardiovascular disease. Although not currently a diagnostic criterion, IR is a cardinal pathophysiological feature and highly prevalent in women with PCOS. Androgens play a bidirectional role in the pathogenesis of IR, and there is a complex interplay between IR and androgen excess in women with PCOS. Skeletal muscle has a key role in maintaining metabolic homeostasis and is also a metabolic target organ of androgen action. Skeletal muscle is the organ responsible for the majority of insulin-mediated glucose disposal. There is growing interest in the relationship between skeletal muscle, androgen excess and mitochondrial dysfunction in the pathogenesis of metabolic disease in PCOS. Molecular mechanisms underpinning defects in skeletal muscle dysfunction in PCOS remain to be elucidated, but may represent promising targets for future therapeutic intervention. In this review, we aim to explore the role of skeletal muscle in metabolism, focusing particularly on perturbations in skeletal muscle specific to PCOS as observed in recent molecular and in vivo human studies. We review the possible role of androgens in the pathophysiology of skeletal muscle abnormalities in PCOS, and identify knowledge gaps, areas for future research and potential therapeutic implications. Despite increasing interest in the area of skeletal muscle dysfunction in women with PCOS, significant challenges and unanswered questions remain, and going forward, novel innovative approaches will be required to dissect the underlying mechanisms.
Introduction
Polycystic ovary syndrome (PCOS) is a common and complex metabolic disorder of women, with a prevalence of 8–13%. 1 It is clinically defined by ovulatory dysfunction, androgen excess (clinical/biochemical) and polycystic ovarian morphology on ultrasound. Androgen excess presents clinically in women with acne, hirsutism or androgenic alopecia, and biochemically is confirmed by the elevation of serum testosterone or other androgen precursors. 2 Insulin resistance (IR) and androgen excess are the cardinal pathophysiological features that underpin the clinical phenotype in PCOS, and are closely correlated clinically and biochemically. 3 The precise direction of causality is unclear, however, and PCOS remains a poorly understood condition despite its high prevalence.
PCOS is increasingly recognized as a primarily metabolic disorder, and it is estimated that approximately 75% of women with PCOS have IR. 4 It is hypothesized that skeletal muscle IR is a major pathophysiological player in driving the adverse metabolic phenotype in PCOS.5,6 Studies have reported reduced insulin-mediated glucose disposal (IMGD) in both lean7–9 and obese women with PCOS.10,11 In this review, we examine the role of skeletal muscle in metabolism, with a particular focus on perturbations in muscle function in PCOS. We also discuss a possible role for androgens in the pathophysiology of observed skeletal muscle abnormalities in PCOS and outline future potential research directions in this regard.
Skeletal muscle structure and function
Skeletal muscle mass compromises 40% of the total body mass and therefore is quantifiably the most abundant tissue mass in humans. 12 Skeletal muscle accounts for 30% of the metabolic rate in adults.12,13 Structurally, there are a number of fibre types that are defined as either slow or fast twitch based on their contractile properties. Classification of muscle fibre type also corresponds with histochemical staining for myofibrillar (myosin) ATPase as type I (slow-twitch) or type II (fast-twitch with highest ATPase activity). Oxidative metabolism is the highest in type 1 fibres, and they stain red because of the abundance of the oxygen transport protein myoglobin which is also related to the mitochondrial density. 14 Similar to adipocytes, muscle fibres are heterogeneous but stable populations that can be regulated by hypertrophy and atrophy and even interconversion. 15 Skeletal muscle is also a metabolic target of androgens. Androgens are known to increase muscle mass in both men and women, and are also associated with visceral adiposity in both sexes.16,17
Skeletal muscle energy metabolism
Seminal work by DeFronzo and others has highlighted the critical role of skeletal muscle in metabolic homeostasis and glucose metabolism.5,18,19 Skeletal muscle is the predominant site of glucose disposal under insulin control; termed insulin-mediated glucose disposal. Skeletal muscle accounts for 80% of IMGD. 20 Skeletal muscle is also the largest store of glycogen in the body and primary site for carbohydrate and lipid metabolism for energy production. 21 Skeletal muscle is responsible for the bulk of fuel oxidation in the body, and has a striking capacity to rapidly modulate the rate of energy production, blood flow and substrate utilization adapting to numerous conditions, including the hormonal milieu and exercise, in both fed and fasted states. 22 Notable enzymes include hexokinase II which phosphorylates glucose, glycogen synthase which controls glycogen synthesis, phosphofructokinase (PFK) which regulates glycolysis and pyruvate dehydrogenase (PDH) which regulates glucose oxidation. 14 For this reason, there is an abundance of mitochondria, as muscle is heavily reliant on oxidative phosphorylation (OXPHOS) for energy production to maintain a continuous supply of ATP. Availability of ATP is modulated to accommodate increased need in times of increased demand, including exercise, in anaerobic conditions (phosphocreatine breakdown and glycogenolysis for substrate-level phosphorylation) and aerobic conditions (OXPHOS by utilizing reduced metabolites of carbohydrate and fat).14,23,24
Regulation of metabolism occurs through inter-organ crosstalk. Communication between adipose tissue, hepatocytes and skeletal muscle maintains metabolic homeostasis. Lipid metabolism and glucose homeostasis are highly interconnected as skeletal muscle utilizes both glucose and free fatty acids (FFAs) as fuel sources.25,26 The role of skeletal muscle metabolism in terms of glucose metabolism, fatty acid (FA) metabolism and the role of mitochondria will be further discussed below and graphically represented in Figure 1.
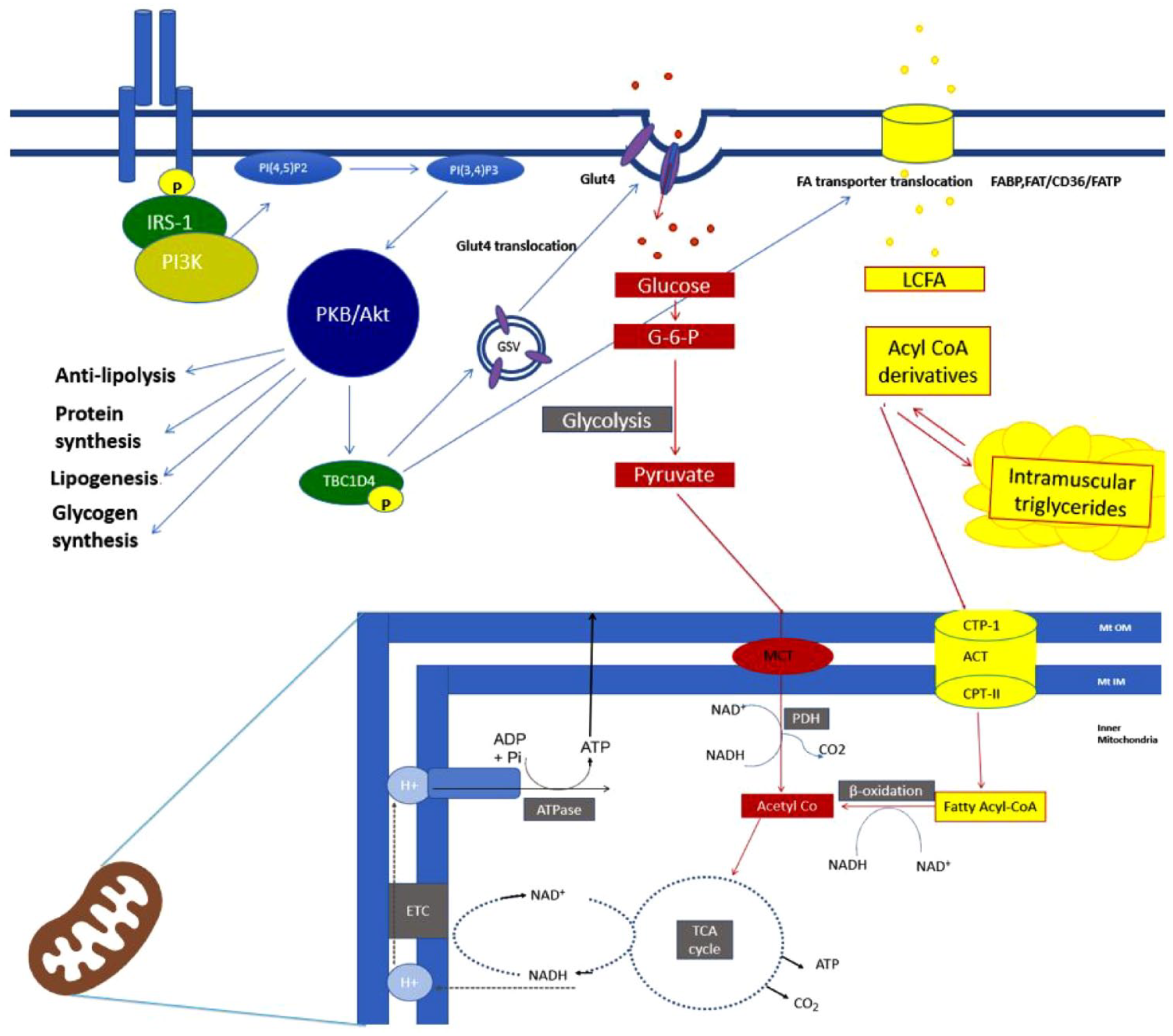
Key metabolic pathways in skeletal muscle. Insulin binding triggers auto-phosphorylation of the insulin receptor and subsequent auto-phosphorylation of IRS-1. Phosphatidylinositol 3-kinase (PI3-K) activation follows, which is a critical insulin signalling node. Akt activation–induced activation of TBC1D4, also known as AS160, initiates GLUT4 translocation to the plasma membrane. Insulin also stimulates the translocation of LCFA transporters to the plasma membrane, a downstream action of Akt, which promotes FA uptake by skeletal muscle while inhibiting FA release at adipocytes. Not shown here is that insulin stimulates protein synthesis effect through mTORC1 activation and inhibits protein breakdown mediated through FoxO.
Key metabolic pathways
As shown in Figure 1, insulin binds to the tyrosine kinase transmembrane receptor which triggers auto-phosphorylation of the insulin receptor and insulin receptor substrate-1 (IRS-1). Phosphatidylinositol 3-kinase (PI3-K) activation follows, which is a critical insulin signalling node. One of the pathways for GLUT4 (glucose transporter 4) translocation is highlighted in this figure through Akt activation–induced activation of TBC1D4 (TBC1 domain family member 4), also known as Akt substrate 160 (AS160). Critical nodes in insulin signalling have been identified as the IRS, PI3-K and the Akt/protein kinase (PKB). 27
GLUT4 translocation to the plasma membrane allows for insulin-mediated glucose uptake into the myocyte. During insulin-stimulated states or fed states, most of the glucose entering the cells is stored as glycogen through the activation of glycogen synthase (GS). Insulin also stimulates protein synthesis effect through mTORC1 activations and inhibits protein breakdown through Forkhead box protein (FoxO) (not shown in Figure 1).
In contrast to the insulin-stimulated state, the fasted state relies on FFA for ATP synthesis. Long-chain fatty acids (LCFAs) are hydrophobic and can cross the plasma membrane, but there is now evidence of the role of membrane-associated FA-binding proteins. Insulin, through downstream activation of Akt, promotes FA update by skeletal muscle through the translocation of LCFA transporters to the plasma membrane and peripherally inhibits FA release at adipocytes. The metabolic status of the myocyte determines the fate of FFA. FFA can be stored once intracellular to monoglycerides, diacylglycerides (DAG) and then triglycerides (TAG) that are known as intramyocellular triglycerides (IMTGs). 28 Excessive lipid flux into skeletal muscle can influence the accumulation of these lipid intermediates which correlates with reduced insulin sensitivity (IS). 29 Of these, diacylglycerol (DAGs) and ceramides are considered most metabolically active and implicated in potential muscle lipotoxicity.22,29–31
The cellular powerhouse, the mitochondria, generates ATP through first β-oxidation of Acetyl-CoA which is an essential substrate for the common final pathway of fuel oxidation, OXPHOS, through the tricarboxylic acid (TCA) cycle. 32 Mitochondrial dysfunction is of interest in metabolic disease, and there is much debate whether this is cause or consequence of IR, type 2 diabetes or obesity. Studies have shown a multitude of perturbations in mitochondrial function including increased reactive oxygen species leading to oxidative stress and mitochondrial damage and depletion, reduced mitochondrial size, downregulated OXPHOS pathways and a decrease in peroxisome proliferator coactivator 1a (PGC1a) activity, which is a master regulator of mitochondrial metabolism. 33
In skeletal muscle, IR manifests as reduced insulin-mediated glucose uptake in the postprandial state and may be present decades before overt hyperglycaemia occurs.18,34 Skeletal muscle function is integrally linked with clinical and metabolic perturbations specific to PCOS, with inter-organ crosstalk graphically demonstrated in Figure 2.
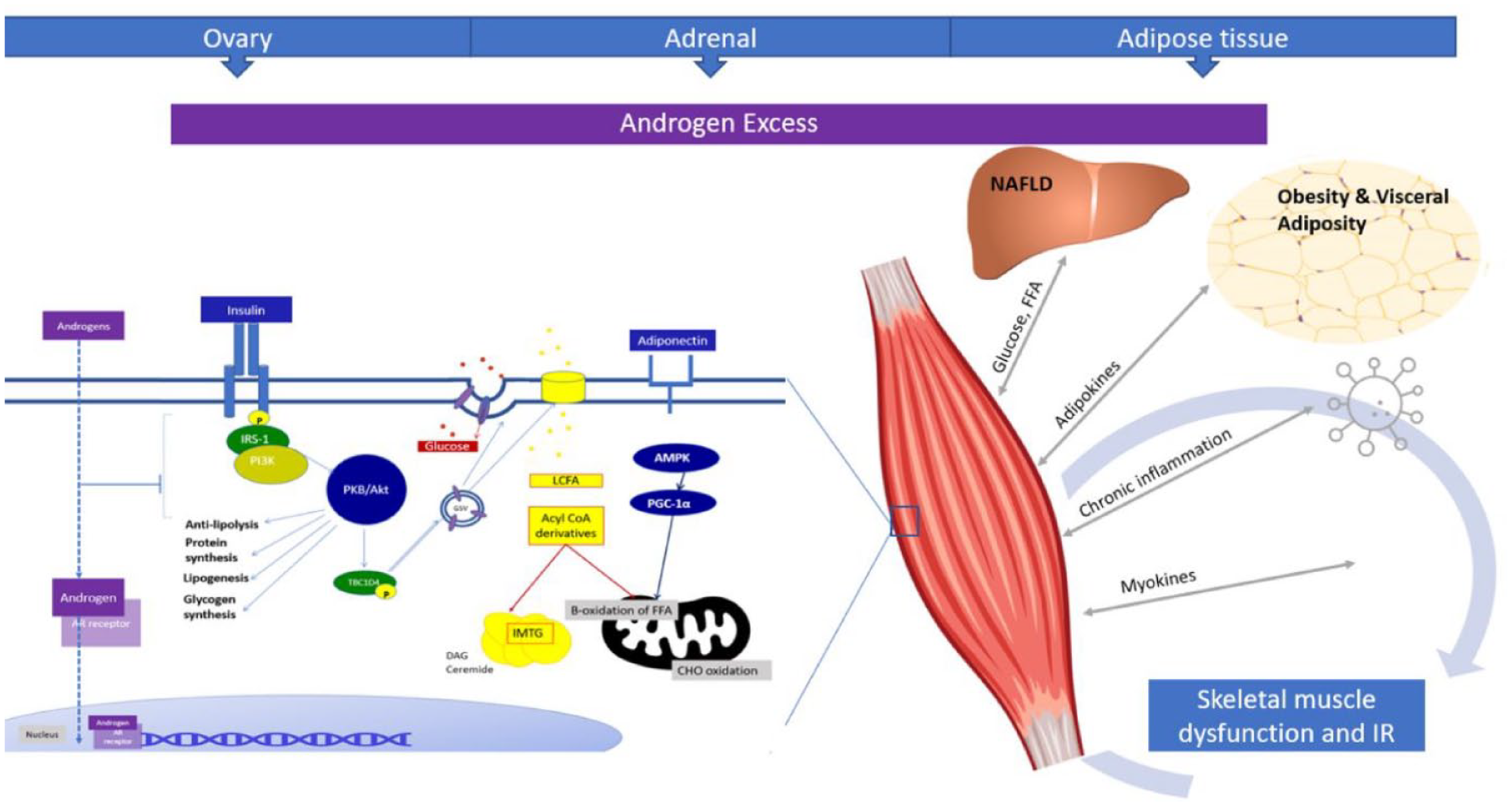
Proposed pathway disturbances in the pathogenesis of PCOS-related skeletal muscle dysfunction. Inter-organ crosstalk highlighting liver, adipose tissue, myokines, chronic inflammation and androgen excess with likely bidirectional implications for metabolic perturbations in PCOS. Intracellular androgen receptor binding with nuclear translocation of the androgen-bound AR receptor demonstrated.
Skeletal muscle dysfunction in PCOS
Poor metabolic health is recognized across the lifespan of women with PCOS.7,34,35 IR and PCOS are both independent risk factors for developing type 2 diabetes.36–41 In PCOS, there is a well-established link between IR, impaired glucose tolerance, type 2 diabetes, non-alcoholic fatty liver disease (NAFLD) and cardiovascular disease (CVD).39,42–44 Circulating androgen burden is associated with an adverse metabolic phenotype, and correlates closely with indices of IR such as Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) and insulin levels. 45 A large-scale genome association study with participants from the UK Biobank has also recently demonstrated that the risk of type 2 diabetes is increased with increasing circulating testosterone concentrations. 46 Perturbations in skeletal muscle energy metabolism appear to be a major player in mediating the adverse metabolic phenotype; understanding the molecular mechanisms underpinning PCOS-specific muscle dysfunction is therefore of critical importance.
IR and PCOS
Postprandial dysglycaemia is more common in women with PCOS than fasting dysglycaemia, and is reflective of a defect in insulin-mediated glucose uptake.47,48 Approximately, 75% of women with PCOS have IR, but this estimate varies across the literature. 49 The variation in reported prevalence is likely a result of a number of factors: method utilized to assess IR, the population studied and also the changing diagnostic criteria for PCOS used in these studies.34,50 For example, different ethnicities may have a higher predisposition to IR. 51 The changing definitions of PCOS over time also affect the interpretation of these phenotyping studies as the diagnostic criteria used introduce heterogeneity in the studied population of PCOS phenotype. 52
Obesity has a strong association with IR, and every body mass index (BMI) increment raises the risk of T2DM by 10% for women with PCOS.53,54 While PCOS is associated with obesity in 50–60% of cases, IR is observed independent of BMI in this condition; IR and risk of diabetes are observed in both lean and obese women. Women with PCOS have significantly decreased in vivo–stimulated glucose disposal indicating IR.7,9 Fasting and dynamic measurements of IS indicate resistance in women with PCOS.9,55,56
This decreased in vivo glucose disposal in PCOS women has been found to be independent of obesity or any alterations in fat-free mass.9,49,57 A systematic review of clamp studies in PCOS highlighted a 27% decrease in IS in PCOS patients, independent of BMI. This review included 28 studies with 38 study estimates, totalling 1224 participants with PCOS compared with 741 controls. 49 Cassar et al. 49 in this meta-analysis also found a relationship between total testosterone and IR, irrespective of the BMI.
Specific defects in insulin signalling in PCOS
Insulin signalling defects are recognized as proximal or distal in respect to Akt, with proximal defects understood as upstream of Akt. Earlier work on insulin signalling was carried out in cultured skin fibroblasts isolated from skin of women with PCOS. 58 This research led to the hypothesis that proximal insulin signalling was impaired through increased phosphorylation of serine residues of the insulin receptor because of a serine kinase which limits progression of insulin signalling beyond the proximal protein of IRS1/2, although this abnormality was not universal in all samples.58–60 Dunaif et al., 61 investigating intact muscle in contrast to cultured myoblasts and adipose culture, demonstrated increased expression of IRS-2 but no change in expression IRS-1. In muscle, IRS-1 leads to GLUT4 translocation, whereas IRS-2 regulates downstream lipid metabolism.21,62,63 While there was no difference in the abundance of the insulin receptor or IRS-1 compared with muscle from controls in the Dunaif study, there was a significant decrease in the IRS-1-associated PI3-K activity in the same paper. 61 Small reductions in the IRS-1 PKB response to insulin in intact muscle were also demonstrated by Hojlund et al.; 64 interestingly, this signalling abnormality was reversed by treatment with insulin-sensitizing medications.
The IRS–PKB pathway is predominantly required for the metabolic effects of insulin such as glucose transport, gluconeogenesis and glycogen synthesis. The mitogenic effects of insulin are mitigated through the RAS-ERK (extracellular signal–regulated kinase) pathway. Corbould et al. 65 have highlighted increased basal phosphorylation of ERK in skeletal muscle of women with PCOS compared with controls, and this trend towards increased basal phosphorylation of ERK was also in agreement with a study by Rajkhowa. 66 Rajkhowa demonstrated that insulin reduced in vivo ERK activity. 66
Hansen et al. 67 have demonstrated perturbations in other glucose-regulating pathways. They focused on intrinsic PCOS factors and, in an attempt to avoid the confounding effect of obesity on IR, studied lean women with and without PCOS. Reduced expression and activity of AMPK (adenosine monophosphate–activated protein kinase) was found. 67 AMPK is a key regulator in the uptake of glucose into cells; AMPK is also linked to reduced circulating adiponectin levels. 67 This study also highlighted reduced metabolic flexibility in response to insulin, linked to reduced PDH. 67
In PCOS, many treatments have a focus on improving IS. Metformin, a biguanide, is commonly prescribed for women with PCOS and is a first line in the treatment of diabetes. Its exact mechanism of action has remained elusive, but interestingly it has been shown that it acts in an AMPK-dependent manner to modulate insulin-mediated GLUT4 translocation. Metformin induces Rab4 expression via AMPK-AS160-PKC to increase insulin-mediated uptake, and AMPK knockdown inhibited this effect. 68 Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with PCOS has been shown to be partially but not completely reversible with peroxisome proliferator–activated receptor (PPAR) alpha therapy in the form of pioglitazone. 64
Exercise is a core lifestyle recommendation for women with PCOS. 69 Exercise improves IS, insulin action and associated with increased skeletal muscle glucose uptake. Exercise improves IS in women with PCOS but does not normalize it.70,71 AMPK also plays a role in regulating glucose uptake during exercise; however, its role is not completely defined. 72 Much of the available data about skeletal muscle metabolism during exercise are derived from studies in males. Females have a higher proportion of type 1 fibres. Females also have a higher reliance on IMTG during exercise which may be the reason women have higher IMTG content in type 1 fibres, with a lower capacity of glycolytic enzymes and a higher reliance on fat during exercise. 73 This effect has been linked to oestrogen levels.73–75
In women with PCOS, Hansen et al. demonstrated that women with PCOS had an impaired ability to upregulate glucose handling proteins for insulin-mediated glucose uptake in muscle. Exercise improved IS in controls by increasing GLUT4 and HK2 gene and protein expressions, but this did not occur in PCOS women. 76
In a recent review, Stepto et al. have highlighted that advances in molecular mechanisms interrogating the perturbations in skeletal muscle in PCOS have to date created more questions than answers. 35 Dissecting mechanistic perturbations to date is hindered by the fact that most of the results are associative rather than causative. 35 Investigating intact muscle perturbations through in vivo studies is challenged by recruitment, expense and differing phenotypes of PCOS, against a background of important confounders such as cardiorespiratory fitness, obesity and physical activity.49,35
Metabolic inflexibility
The capacity of an organism to switch between oxidizing fat in the fasted state to carbohydrate in the fed state is termed metabolic flexibility. 22 Metabolic inflexibility is well-documented in the setting of obesity, IR and type 2 diabetes. Its association with PCOS is also well-characterized.77–79
While the molecular basis remains uncertain, it has been postulated that metabolic inflexibility implies the inability to appropriately activate glucose oxidation in response to glucose load; this may simply reflect a secondary consequence of impaired insulin action. In contrast to this theory, defects in mitochondrial fuel selection, independent of insulin signalling, could be a primary driver of IR, or it may occur as a result of oversupply of lipid fuel. 80 Metabolic inflexibility can lead to lipid accumulation in ectopic tissues such as skeletal muscle, driving lipotoxicity and exacerbating IR. In women with PCOS, ceramide has been increased threefold in one study with an overall 1.5-fold increase in IMTG compared with healthy controls. 67
Metabolic inflexibility in PCOS is more pronounced with high BMI, worsening IR and hyperandrogenaemia.81,82 A recent systematic review has highlighted the association with hyperandrogenism, although the pathogenesis is unclear. 82 There appears to be a common pathway in PCOS for inappropriate energy oxidation, metabolic inflexibility and impaired response to insulin in women with PCOS, but the role of androgen excess remains to be elucidated.82,83
The role of androgen excess in skeletal muscle dysfunction
A direct causal relationship between androgen excess and IR in PCOS has been explored in a number of in vivo studies, with a focus on the effect of androgens on IS and whole-body glucose uptake through euglycaemic-hyperglycaemic clamp studies. 49 Abnormal insulin signalling is undoubtedly a feature of National Institutes of Health (NIH) and Rotterdam PCOS phenotypes with androgen excess, and may conceivably be milder in non-hyperandrogenic cohorts. In the face of changing diagnostic criteria over time, abnormal insulin action is also still recognized in those with PCOS who have hyperandrogenism, polycystic ovaries and seemingly normal ovulatory function. 84 This IR may be milder than those with ovulatory dysfunction. 84 A recent population study has shown that women with PCOS exposed to combined oral contraceptive pills (COCPs) had a reduced risk of developing dysglycaemia across all BMI subgroups, raising the possibility of modulation of androgen action as a therapeutic tool to reduce metabolic dysfunction in PCOS. 85
Testosterone exposure can be amplified within target tissues. T is reduced by the intracellular enzyme 5-alpha reductase to dihydrotestosterone (DHT). DHT is considerably more potent at the androgen receptor than testosterone. 86 Labrie et al. 87 in 1988 coined the term intracrinology, which describes this mechanism of hormone precursor activation, action and inactivation within the peripheral target cells. At a cellular level, skeletal muscle cells exhibit intracellular 3-beta hydroxysteroid dehydrogenase (HSD), 17-beta-HSD and 5-alpha reductase activity resulting in the activation of dehydroepiandrosterone (DHEA) to T and DHT. 86 AKR1 C3 (aldo-keto reductase type 1 C3), also known as 17 beta-HSD type 5, is an androgen activating enzyme, converting androstenedione (A4) to T, and has been proposed as an essential enzyme of androgen activation in skeletal muscle.88–91 Pre-receptor androgen metabolism has been extensively characterized in adipose tissue, but to date there is a relative paucity of data on androgen activation in skeletal muscle. 86
There are a number of in vivo studies examining the role of androgen exposure on IS in women to understand a potential causative role for androgens in the pathogenesis of IR. Utilizing the hyperglycaemic-euglycaemic clamp, a significant reduction in whole-body IS has been demonstrated when short-term androgens were administered to women with normal menstrual cycles and BMI. 92 Clamp studies were completed before and after at least 10 days of methyltestosterone. 92 In one study, testosterone administration to patients undergoing female-to-male sex reassignment resulted in a significant decrease in IMGD measured by euglycaemic clamp studies before and after 4 months of treatment. 93 Zang et al. studied testosterone administration to postmenopausal women before and after 3 months. They randomized women with average BMI 24.1 kg/m2 to one of three groups: testosterone undecanoate, estradiol valerate or both. IS was significantly reduced in any of the testosterone groups without change in the oestradiol group. 94
Studies of anti-androgen treatment in hyperandrogenic women have also been shown to improve IS. Moghetti et al. implicated androgens in IR by highlighting the impact of androgen receptor blockade on IS before and after 3 months of anti-androgen treatment. They used three different anti-androgen treatments: spironolactone, flutamide and the gonadotropin-releasing hormone (GnRH) agonist buserelin to block androgen action. With all anti-androgen treatments, there was significant improvement in IS as measured by clamp in lean hyperandrogenic women before and after 3 months. 95
Intriguingly, 11-oxygenated androgens represent the majority of circulating androgens in women with PCOS, and these androgens do not decline with age in comparison with classic androgens and remain elevated after menopause. 96 The question of whether these androgens contribute to metabolic risk and IR in PCOS is unresolved. The 11-oxygenated precursor androgens β11-hydroxyandrostenedione (11OHA4) and 11-ketoandrostenedione (11KA4) have been found to correlate with BMI, fasting insulin and HOMA-IR, but study results are conflicting and the same relationship with 11-ketotestosterone (11KT) and 11-hydroxytestosterone (11OHT) has not been found. Recently, Tosi et al. 97 describe a positive relationship between 11-oxygenated androgens and IS, suggesting a relationship with IS that may diverge from that of classic androgens. This highlights the importance of future research to dissect these observations further.
Other mechanisms of PCOS-related muscle dysfunction
Myokines and PCOS
Skeletal muscle is increasingly recognized as an endocrine target organ. 98 Myokines are defined as cytokines and other peptides that are produced and released by muscle and exert autocrine, paracrine or endocrine effects. 99 Adiponectin, while a classical adipokine, is also now recognized as a myokine secreted from muscle with autocrine and paracrine effects. 100 Its effect on skeletal muscle includes metabolically favourable roles in the regulation of IS, metabolism, protein turnover, inflammatory signalling and myogenesis. 101 Lower adiponectin levels have been associated with lower AMPK Thr172 phosphorylation in skeletal muscle. 67 High-molecular-weight (HMW) adiponectin is inversely associated with intramyocellular lipid content detected by proton magnetic resonance spectroscopy in skeletal muscle. 102 Adiponectin receptor 1 (AdipoR1) has a crucial role in skeletal muscle; adiponectin and AdipoR1 induce PGC-1a activation through Ca2+ signalling and by AMPK which leads to increase mitochondrial biogenesis. 103 After controlling for BMI-related effects, adiponectin levels are lower in women with PCOS compared with non-PCOS controls, likely related to IR. 104 Androgen exposure can also cause a reduction in total circulating adiponectin by its inhibition of secretion from adipocytes.104–106
Irisin, a myokine discovered in 2012, is typically considered to have a positive association with energy metabolism; it is upregulated by exercise and downregulated in metabolic disease. 107 Its role in PCOS has recently attracted attention. Zhang et al. 108 demonstrated elevated irisin levels in women with hyperandrogenism and IR, while irisin levels were normal in women with PCOS without androgen excess. They postulated that elevated irisin levels may increase in a compensatory mechanism to metabolic stress at a molecular level in an attempt to counterbalance IR in women with hyperandrogenism and PCOS. 108 Li et al. also demonstrated irisin to be elevated in women with PCOS and androgen excess after adjusting for confounders such as BMI. Circulating irisin levels predicted hyperandrogenism, metabolic syndrome and IR in women with PCOS. 109 Overall, a meta-analysis in 2018 of eight studies did not observe any significant correlation between circulating irisin levels and IR in the PCOS population studied, and further studies are required to conclusively address this issue. 110
Myostatin, a myokine belonging to the transforming growth factor beta (TGF-β) superfamily, negatively regulates muscle mass and growth. Myostatin also decreases insulin signalling and glucose uptake causing IR through nuclear factor kappa B (NF-κB). 111 Inhibition of NF-κB in vitro improves IR in L6-cultured muscle cell exposed to H2O2. 112 Chen et al. 113 found that myostatin correlated with abdominal obesity and was increased in overweight women with PCOS. Further studies have not identified a difference in myostatin levels in women with or without PCOS. 114 Obesity per se is associated with increased myostatin expression, and this has been demonstrated in muscle biopsies. 115
Muscle mitochondrial dysfunction in PCOS
Abnormal mitochondrial function can affect whole-body metabolic homeostasis. Oxidative stress is considered to be a major contributor to IR in PCOS. Obesity is also a cause of oxidative stress; however, non-obese women with PCOS have increased oxidative stress, and therefore, this does not appear to be mediated by obesity alone. 116 Reactive oxygen species–induced oxidative stress may also induce a pro-inflammatory state contributing to IR.117,118
In women with IR and PCOS, genes involved in mitochondrial oxidative metabolism were downregulated in skeletal muscle. 119 Pioglitazone-induced improvements in IS were demonstrated in the same study cohort with increased expression of nuclear-encoded genes in skeletal muscle involved in mitochondrial phosphorylation pathways, OXPHOS. Pioglitazone increased the expression of PGC-1a in muscle of PCOS patients in this study cohort, and they postulated that this may occur via an increase in adiponectin which is an effect of thiazolidinedione (TZD) on muscle PPAR-y and an improved insulin action on mitochondrial biogenesis.120,121
The PCOS muscle transcriptome and emerging role of microRNA
Women with PCOS have altered skeletal gene expression that is associated with specific changes in skeletal muscle DNA methylation patterns. 122 Epigenetic changes are of increasing interest given genome-wide associated studies only explain a modest proportion of the heritability of PCOS, which was estimated at 70% from twin studies.123,124 Transcriptomic and epigenetic analysis show promise in delineating the perturbed molecular mechanisms in skeletal muscle IR in PCOS.
Nilsson et al. 122 highlighted aberrant gene expression in a number of genes related to lipid, glucose metabolism and circadian rhythm that may be involved in the metabolic abnormalities of PCOS. Decreased COL1A1 expression correlated negatively with IR and interestingly COL1A1 expression may be explained by hyperandrogenism in women with PCOS. 122 Testosterone exposure in vitro decreased COL1A1 expression in myotubes. This group also found that insulin regulated gene expression of Krüppel-like factor 10 control element (KLF10 or TGF-β-inducible early growth response protein 1). This is a DNA-binding protein which is already shown to be implicated in hepatic fibrosis via its binding with TGF-β control element.125,126
There is increasing interest in skeletal muscle fibrosis and the aberrant gene expression in pro-fibrotic pathways that have been demonstrated by Stepto et al. 127 The implication of these pathways in skeletal muscle IR is unknown but has been highlighted as a plausible contributor to IR; it is hypothesized that TGF-β ligand–mediated stromal deposition or fibrosis may be implicated in skeletal muscle in women with PCOS. This group demonstrated elevated gene expression in the TGF-β-regulated tissue fibrosis pathway. Specifically, changes have been noted in genes that encode extracellular matrix (ECM) components (COL1A2, COL3A1), enzymes in collagen deposition and assembly (LOX, DCN), ligands (TGFB2) and their receptor. 127 Functional studies or proteomic analysis may advance this hypothesis in the future. 128
Furthermore, in the study by Nilsen et al., COL1A1 was associated with increased DNA methylation of a CpG; DNA methylation at only two sites was highly correlated with differential gene expression in this particular study. The most overexpressed gene was noted to be DYRK1A that encodes an inhibitor of glycogen synthase kinase-3; this was not affected by in vitro androgen exposure, but 3 days of in vitro insulin exposure did reduce the cell response to insulin. 122 NAMPT, a gene encoding visfatin, was upregulated in women with PCOS. Visfatin may have a role in mitochondrial biogenesis and has also been show to play a role in skeletal muscle uptake via the Ca2C-mediated AMPK–p38 mitogen-activated protein kinase (MAPK) pathway. 129
Utilizing transcriptional profiling, it has been demonstrated that IR in skeletal muscle of women with PCOS is associated with altered gene expression. The authors reported reduced expression of genes involved in mitochondrial oxidative metabolism and postulated that reduced expression of peroxisome proliferator–activated receptor-γ coactivator (PGC)-1αP could be implicated in this abnormality. 119 PGC-1α is a key regulator of mitochondrial biogenesis and also modulates remodelling of muscle to fibre-type composition, as well as participating in carbohydrate and lipid metabolism. Skov et al. 119 indicated that these transcriptional abnormalities may contribute to the unique phenotype of IR in patients with PCOS. Exercise has positive effects on gene expression and epigenetic profiles in skeletal muscle of those with IR. 130 Of interest, a study highlighted that electroacupuncture alters DNA methylation and could reverse PCOS-related transcriptomic disturbances in skeletal muscle, inducing metabolically advantageous alternations similar to those observed with exercise. 131
There is also increasing interest in microRNA (miRNA) and PCOS development, and it is postulated that miRNA expression might be involved in the underlying pathophysiology of PCOS including glucose metabolism and IS. 132 miRNA is short non-coding RNA molecules that are widely present in different tissue environments. MicroRNA plays critical role in gene regulation at post-transcription level and shows promise as potential biomarkers. Jing et al. 133 have identified three miRNAs (mrR-122, mrR-193b and miR-194) that are increased in women with PCOS and impaired glucose tolerance; there was also a modest correlation with HOMA-IR.
Conclusion
Although PCOS-specific mechanisms of skeletal muscle metabolic dysfunction have been identified, there is unlikely to be a single unifying molecular defect that underpins the adverse metabolic phenotype in this heterogeneous and complex disorder. Crosstalk between multiple contributory factors – including genetic, environmental, obesity and hormonal, coupled with intrinsic insulin signalling defects – is likely central to conferring the adverse phenotype observed. The role of androgen excess as a primary driver of some of these perturbations remains to be elucidated, and there is increasing interest in mitochondrial dysfunction as a potentially primary rather than secondary factor. Understanding the role played by androgen excess in PCOS-related skeletal muscle dysfunction, as well as the differential impact of classic and 11-oxygenated androgens in these processes, should be the focus of future research efforts. Novel and innovative approaches will be required to delineate these relationships.
Given the central role of skeletal muscle in IMGD, ameliorating IR at this site may effectively improve whole-body glucose homeostasis and may help abate the vicious cycle of hyperinsulinaemia and hyperandrogenaemia that is evident in PCOS. Future investigation into the role of skeletal muscle in PCOS and better understanding of the molecular basis may lead to future development of novel targeted therapies. The advent of ‘omics’-based approaches such as genomics, transcriptomics, proteomics and metabolomics in large phenotyping studies may be helpful to further our understanding of skeletal muscle dysfunction and skeletal muscle IR in PCOS. It remains unclear whether modulation of defects in skeletal muscle energy metabolism, or indeed of local androgen exposure in muscle tissue, will live up to its potential as a novel target to reduce metabolic risk.