
Abstract
Polycystic ovary syndrome (PCOS) has been traditionally perceived as a reproductive disorder due to its most common presentation with menstrual dysfunction and infertility. However, it is now clear that women with PCOS are at increased risk of metabolic dysfunction, from impaired glucose tolerance and type 2 diabetes mellitus to nonalcoholic fatty liver disease and cardiovascular disease. PCOS is characterised by androgen excess, with cross-sectional data showing that hyperandrogenism is directly complicit in the development of metabolic complications. Recent studies have also shown that C11-oxy C19 androgens are emerging to be clinically and biochemically significant in PCOS, thus emphasising the importance of understanding the impact of both classic and C11-oxy C19 androgens on women’s health. Here we discuss androgen metabolism in the context of PCOS, and dissect the role played by androgens in the development of metabolic disease through their effects on metabolic target tissues in women.
Introduction
Polycystic ovary syndrome (PCOS) is one of the most common endocrine conditions, affecting approximately 10% of all women across the globe. 1 Women with PCOS commonly present with menstrual irregularities, infertility and signs and symptoms of androgen excess. PCOS has been traditionally perceived as a predominantly reproductive disorder, with minimal focus on long-term metabolic complications. However, recent studies have shown that PCOS is associated with significant health consequences in affected women, 2 representing a lifelong metabolic disorder (Figure 1).

Iceberg phenomenon: PCOS has been traditionally perceived as a primarily reproductive disorder. On a superficial level, clinical consequences of this condition include subfertility, irregular menses and signs and symptoms of androgen excess. However, clinicians should be aware of the significantly increased risk of metabolic complications across the female lifespan in women with PCOS, underpinned by androgen excess and insulin resistance.
Although controversies persist with regard to diagnostic criteria for PCOS, androgen excess remains a fundamental biological and diagnostic feature of the disorder.3,4 This is recognised in the 2003 Rotterdam Consensus criteria, with androgen excess featuring as a defining criterion alongside irregular periods and polycystic appearances of the ovaries at ultrasound. Following this, the Androgen Excess Society recommended hyperandrogenism as a mandatory criterion for the diagnosis of PCOS, further highlighting PCOS as a disorder of androgen excess.4–6 In cross-sectional clinical phenotyping as well as population studies, androgen excess is a major driver of metabolic risk in PCOS, with the presence and severity of androgen excess closely correlating with surrogate parameters of metabolic risk, including insulin resistance. 7 Women with PCOS have been shown to be at increased risk of metabolic disease including type 2 diabetes mellitus (T2DM), nonalcoholic fatty liver disease (NAFLD), and cardiovascular disease (CVD). 8
Most studies on PCOS have focused on classic C19 androgens, their precursors and/or their downstream urinary metabolites for biochemical assessment of androgen excess. However, the newly described, adrenal-derived C11-oxy C19 androgen subclass is emerging to be clinically and biochemically significant in the context of PCOS-related androgen excess. 9 In our recent study, we found that C11-oxy C19 androgens are the majority of circulating serum androgens in women with PCOS 10 , thus emphasising the importance of improving our understanding of the impact of both classic and C11-oxy C19 androgens on women’s health. Furthermore, circulating levels of the active C11-oxy C19 androgen 11-ketotestosterone (11KT) have recently been shown to remain consistent throughout the female lifespan, 11 contrary to classic androgen concentrations, which decrease post-menopause. However, recent study by Swart et al. 12 analysing steroid hormones of adolescents and young women with PCOS and adrenocortical dysfunction using UPC2-MS/MS found C19 steroids [A4, testosterone and dehydroepiandrosterone (DHEA)] concentrations higher than the combined C11-oxy C19 androgens. The combined C19 steroid levels were higher in PCOS group with the combined C11-oxy androgens being similar in patients with PCOS and healthy people. Therefore, more studies on C11-oxy C19 and their downstream metabolites will be needed before a conclusion can be drawn about the roles of C11-oxy C19 androgens compared with the classic androgens.
Conditions characterised by androgen excess are PCOS, premature adrenarche, congenital adrenal hyperplasia, ovarian hyperthecosis, androgen secreting tumours and, to a degree, also Cushing’s syndrome.13,14 Many of these conditions lack the necessary prevalence and frequency of life events and metabolic dysfunction to understand the overall impact of androgen excess. 15 PCOS is a lifelong metabolic condition likely to affect female health from childhood through to post-menopause. Recent work by Risal et al. showed that daughters of mothers with PCOS have a fivefold increased risk for PCOS. 16 More importantly, they showed that prenatal androgen exposure and not obesity lead to transgenerational reproductive and metabolic dysfunction in rodent models. 17 Another study by Gunning et al. showed subtle cardiometabolic dysfunction in early childhood in otherwise healthy weight children of women with PCOS.18,19 Based on its high prevalence in the general background population, and the correlation between PCOS and other features of metabolic syndrome, PCOS is a good model for studying the impact of androgen excess on metabolic risk.The origins of androgens and their mechanistic role inducing metabolic dysfunction in PCOS are discussed in detail in the following.
Review methodology
We included both in vitro and in vivo human and animal studies published in English up until March 2020 providing evidence on PCOS and metabolic dysfunctions. Articles were searched from PubMed using terms “(PCOS) OR (PCOS MeSH terms)” AND (Androgens) OR “(Androgens MeSH terms)” AND terms on metabolic dysfunctions as mentioned previously such as “Obesity”, “Diabetes” and with their associated MeSH terms. We also added specific organs such as “Adrenal”, “Ovary”, “Muscle”, “Pancreas”, “Liver” in separate searches to review their impact on PCOS and metabolic dysfunctions. Additional articles were also obtained from previously published reviews for respective subjects. Articles were then screened based on titles and abstracts and full texts were subsequently obtained.
Origins of androgen excess in PCOS and pre-receptor androgen metabolism
Androgens are responsible for the development of male characteristics such as male pattern body hair, muscle bulk, and deep voice; androgens also impact on sexual function and reproductive capacity in both sexes. In women, androgens also play an important role in general wellbeing, libido, energy levels, muscle mass and strength and bone mass.20–24 Apart from their role in reproduction and sexual health, androgens also play an important role in human metabolic health in both men and women. 25 The origins of androgen excess in PCOS have not been comprehensively delineated, but adrenals, ovaries and adipose tissue collectively contribute to the bulk of circulating androgen burden in the disorder. 8 Androgens are synthesized in the ovaries and adrenal glands catalysed by a series of steroidogenic enzymes, especially the rate limiting cytochrome P450 (CYP) 17A1 enzyme, responsible for the synthesis of the classic androgen pathway precursor DHEA. DHEA and its downstream product androstenedione are released into circulation, with androgenic signals further amplified after uptake into target tissues, where androgen precursor steroids are converted into the active androgens testosterone and 5α-dihydrotestosterone (DHT), the most potent natural androgen. 26 Several studies have reported a systemic upregulation of 5α-reductase activity as a significant feature in women with PCOS,27–31 with consequently increased activation of testosterone to 5α-DHT. More recently, another pathway of DHT synthesis, referred to as ‘backdoor pathway’ has been discussed in the context of PCOS and congenital adrenal hyperplasia.32–34 This pathway involves the synthesis of DHT bypassing testosterone in the classic pathway. There are also speculations of androgens transferring via mother’s milk into children as an early source of androgens in neonatal and infant life. However, currently there is a lack of scientific evidence to support this theory. As PCOS is a heterogeneous condition associated with features of the metabolic syndrome, in vivo studies involving women with PCOS are often confounded by coexisting obesity and insulin resistance, which are also contributors to hyperandrogenism in women with and without PCOS. 35
Ovaries and androgen excess
The origin of androgen excess from the ovaries is complex and mainly attributed to tonic hypersecretion of Luteinizing Hormone (LH), likely due to a dysregulated hypothalamic–pituitary–gonadal axis. 36 A theory of insulin-potentiated accelerated gonadotropin-releasing hormone (GnRH) pulse frequency, resulting in increased LH pulse is supported by the works of Roland and Moenter.37,38 Insulin has also been shown to stimulate androgen productions in the ovarian theca cells of women with PCOS via insulin receptor rather than insulin growth factor 1 (IGF-1) receptor.39,40 However, the IGF-1 receptor may also play a role in conditions with extreme levels of insulin resistance such as heridatery insulin receptor mutations or lipodystrophy contributing to their hyperandrogenism and PCOS.41,42 High levels of IGF-1 in acromegaly have also been previously associated with PCOS 43 with the observation of improvement in IGF-1 and PCOS phenotype following reduction in growth hormone levels. This was followed by the normalisation of menstrual cycle and numbers of polycystic ovary morphoplogy suggesting that IGF-1 could play a role in hyperandrogenism and PCOS. 44 Low concentrations of progesterone have been proposed to increase LH secretion by disrupting the negative feedback of GnRH. 45 However, the presence of increased LH and androgen excess in prepubertal girls fails to support low progesterone as a sole source of LH increase in PCOS. 46 The increase in ovarian androgens in PCOS is not only LH- and insulin-dependent as some in vitro studies have shown that it could be due to an abnormality in primary theca cell steroidogenesis.47,48 Subsequent clinical studies in women with PCOS have shown that ovarian androgen production is hyperresponsive to both administration of GnRH agonists and Human Chorionic Gonadatropin (hCG) challenges.48–51 The role of upregulation of backdoor androgen synthesis pathway contributing to androgen excess in women with PCOS has been discussed by Marti et al. 52 In their study, they have shown that the ovaries express all the enzymes required for DHT synthesis through the backdoor pathway. Comparing ovarian tissues of women with PCOS to women without PCOS, immunohistochemistry revealed higher reactivity for 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2), retinol dehydrogenase (RoDH), 5α-reductase type 1 (SRD5A1) and aldo-keto reductase family 1 member C2 (AKR1C2) genes indicating a possible higher activities of their respective enzymes involved in the backdoor pathway. SRD5A1, the gatekeeper of the backdoor pathways converting A4 into androstanedione, showed the highest difference in PCOS compared with non-PCOS highlighting the relevance of this pathway in women with PCOS. Derangement in the Kiss-1 system resulting in increased LH release has also been postulated; however, evidence supporting this theory is lacking to date. 36 Recently, the role of Gamma Aminobutyric Acid (GABA) activity in the arcuate nucleus stimulating LH secretion resulting in PCOS-like reproductive dysfunction has been explored in mice by Silva et al. 53
Adrenal glands and androgen excess
Nearly 50% of women with PCOS have a predominantly adrenal hyperandrogenism phenotype as shown by elevated concentrations of Dehydroepiandrosterone Sulphate (DHEAS) and 11β-hydroxyandrostenedione, two androgens that are exclusively produced by the adrenal glands.54–56 This is particularly prominent in young women with PCOS. 13 This may be due to upregulation of CYP17A1 activity through tonic hyperstimulation by Adrenocorticotropic hormone (ACTH) of the adrenal gland. Lack of a significant increase in 17-hydroxyprogesterone (17OHP) after exogenous stimulation with ACTH supports this theory.54,57 One of the other proposed mechanisms of adrenal hyperandrogenism is a relative deficiency of HSD3B2. In women with PCOS and high DHEAS, a normal DHEA response and slightly elevated 17OHP response to ACTH suggested that elevated DHEAS was due to tonic hyperstimulation of the adrenal gland rather than deficiency of the enzymes. 54 Downregulation of 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1) activity will result in a decreased rate of cortisol activation and hence could lead to adrenal androgen stimulation via hypothalamic–pituitary–adrenal axis feedback; however, no convincing evidence for decreased HSD11B1 activity was found in two independent studies in women with PCOS.58,59 The relevance of the adrenal backdoor pathway in women with PCOS was also discussed by Saito et al. showing correlations between the levels of DHEAS to androstanedione and androsterone indicating a potential adrenal gland contribution in these two androgens involved in this pathway. 60
Importantly, the C11-oxy C19 androgens are primarily of adrenal origin as this pathway starts with the conversion of A4 to 11-hydroxyandrostenedione (11OHA4), which can only be catalysed by 11β-hydroxylase activity of the exclusively adrenally expressed enzyme CYP11B1.55,61 As C11-oxy C19 androgens are particularly prominent in the serum women with PCOS 10 as well as in girls with premature adrenarche, 62 it is safe to assume that the adrenal glands provide a major contribution to circulating androgen excess in PCOS (Figure 2).

Adrenal, ovarian and peripheral androgen metabolism in PCOS. Androgenic precursors are secreted predominantly by the adrenal glands, and activated to potent androgens in the ovaries and peripheral tissues. Expression of key androgen-activating enzymes in peripheral tissues such as adipose tissue highlight an important role for extra-adrenal and -ovarian androgen generation.
Peripheral tissues and androgen excess
After the secretion by the ovaries and/or adrenals into the circulation, androgenic precursors are further activated in peripheral target tissues into more potent androgens that bind to the cytosolic androgen receptor (AR), which subsequently translocates to the nucleus where it functions as a transcription factor after binding to DNA, resulting in increased transcription of genes encoding for factors important for androgenic biologic activity. The concentration of androgens in peripheral target tissues of sex steroid action are determined by (1) the total concentration of the androgenic steroids in the circulation (2) whether they are bound to sex hormones binding globulin (SHBG) or albumin (3) important regulators of bioavailability, and the availability of mechanisms for cellular influx and efflux. Circulating androgens must cross the plasma membrane of the target cells to be metabolized by intracellular enzymes and/or to activate AR. While unconjugated steroids can freely diffuse across the plasma membrane, conjugated steroids are hydrophilic and hence need to be actively transported inside the cell. Once in the cell, these steroids will need to be deconjugated before they can be metabolized and/or interact with the androgen receptors.25,63
Combining human-based in vivo and ex vivo study approaches, our group has shown that the androgen-activating enzyme aldoketoreductase type 1 C3 (AKR1C3), which converts androstenedione to testosterone as well as 11OHA4 to 11KT, is a major driver of adipocyte-specific androgen excess and that its activity in adipose tissue is a major source of PCOS-related androgen excess. 64 This will be discussed in detail in the adipose tissue section in the following.
MicroRNA and androgen excess
MicroRNAs (miRNAs) are present widely in the body playing a role in the regulation of gene expression by inhibiting post-transcriptional or post-translational expression of messenger RNA (mRNA). 65 Alteration in levels of miRNA have been implicated in conditions such as cervical cancer, ovarian cancer, endometriosis and CVDs.66–68 Studies have shown differential expression of miRNA in women with PCOS compared with controls, suggesting a potential role of miRNA in PCOS.69,70 Chen et al. have extensively discussed the role of miRNA in PCOS. 71 Muri et al. have previously shown that levels of miR-21, miR-103 and miR-155 were positively associated with levels of free testosterone in women with PCOS 72 and several miRNAs have been shown to be negatively correlated with levels of testosterone and A4 in these women.73,74 Moreover, comparing miR-130b-3p in normal and theca cells of women with PCOS have shown a decreased expression in PCOS which was correlated with increased expression of CYP17A1 and DHEA synthesis, which may contribute to the androgen excess in PCOS. 75
In isolated porcine ovaries, overexpression of miR-378 was shown to inhibit the expression of aromatase enzymes in the ovaries, which contributed to hyperandrogenaemia. 76 Furthermore, miRNA181a also downregulates oestrogen synthesis via CYP19A1 expression in mouse granulosa cells. 77 However, there is no evidence of miRNA overexpression in women with PCOS, hence its role in increasing levels of androgens in this population. miR-193a-5p and miR-199a-3p have been positively associated with oestrogen and SHBG and negatively associated with free testosterone in women with PCOS with predictions of target genes, indicating the role of these miRNA in regulating some enzymes in the steroidogenesis pathways. 78 These evidences taken together suggested that more detailed studies will be needed to investigate the role of miRNA in contributing to androgen excess in women with PCOS. The role of miRNA in propagating metabolic diseases will be discussed in the relevant section in the following.
Androgens in metabolic target tissues
Critical metabolic target organs such as adipose tissue, muscle and pancreatic beta cells are also targets of androgen action (Figure 3). Here, we summarise the potential mechanisms and metabolic dysregulation due to androgen action in each of these organs.
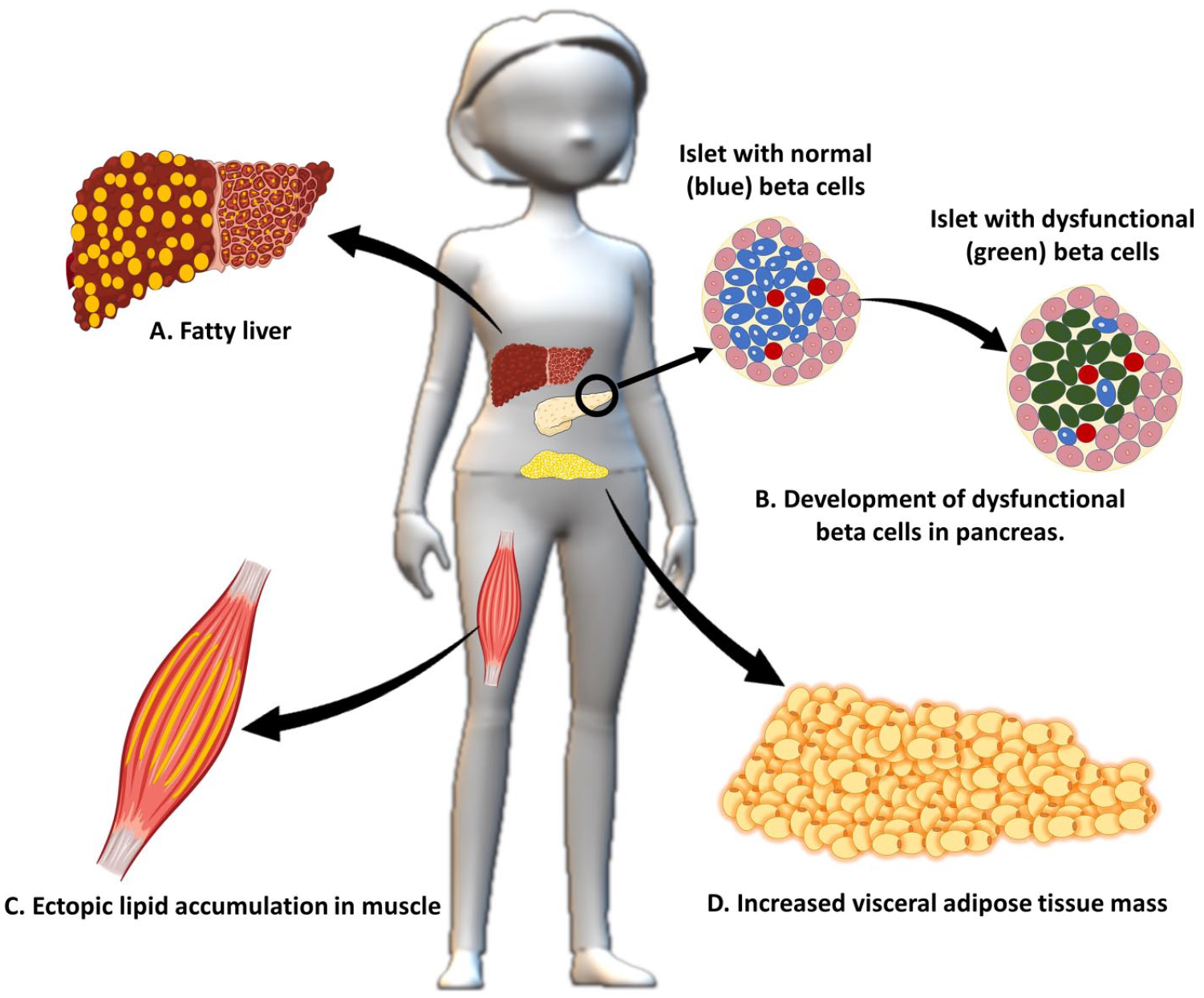
Impact of androgen excess on metabolic target tissues- (A) Increased fat accumulation in the hepatocytes in response to androgen excess, eventually resulting in NAFLD, (B) Androgen excess is proportionally related to beta-cell dysfunction, (C) Ectopic lipid accumulation in skeletal muscle with androgen excess influences glucose-regulating pathways resulting in insulin resistance, (D) under the influence of androgens, women with PCOS have been shown to have more central obesity phenotypes.
Adipose tissue, obesity, fat mass distribution and androgen excess
Adipose tissue forms the largest store of conserved energy, in the form of triglycerides, in the human body. The process of adipocyte differentiation from stem cells is complex and involves a variety of signals for gene regulation, histone modification, and protein modification by ubiquitin. 79 There are two distinct types of adipose tissue, brown adipose tissue and white adipose tissue. Several studies have established the role of adipose tissue beyond an energy storehouse to regulating several critical physiological processes for example, through adipokine signals such as leptin and adiponectin. The various roles of adipokines is discussed in detail by Ouchi et al. and Dimitriadis et al.80,81
Women with PCOS have been shown to have more central obesity phenotypes compared with weight and body mass index (BMI)-matched controls,82–85 which is associated with various metabolic conditions in PCOS.86–88 However, these data have been inconsistent.89,90 Some studies have also shown that although women with PCOS have android fat mass distribution, these might not be accompanied by increased visceral fat.91,92 On the other hand, gluteo-femoral fat or gynecoid adiposity, low in PCOS, has been shown to be independently associated with protective lipid and glucose profile along with decreased cardiovascular and metabolic risk. 93 Visceral adiposity index (VAI) has similar utility to the gold-standard computed tomography scan in the evaluation of visceral adiposity. 94 VAI is a sex-specific empirical mathematical model based on BMI, waist circumference, triglycerides and high-density cholesterol in healthy normal and overweight populations. 95 Lim et al. reported a similar VAI in women with PCOS and healthy controls in few studies. However, subgroup analysis comparing three groups (PCOS and obesity, obesity alone, and PCOS alone) revealed that women with PCOS and obesity have higher VAI compared with the other two groups. 96 In a study on women with PCOS by Amato et al., VAI was only associated with compensatory hyperinsulinemia and there was no independent association between VAI and insulin sensitivity index (by HOMA-IR and ISI Matsuda), androgen profiles and PCOS. 97 Visceral adiposity has also been previously, but not consistently, associated with elevated FAI 82 and total testerosterone. 85 However, the number of studies available is too low for a systematic review to examine whether there is an association between PCOS, central obesity and androgens.
Adipose tissue is a key target of androgen action. Androgens are known to inhibit adipocyte differentiation in both animal and human cell lines.98–101 The effects of androgen on adipose tissue can be noted from birth. Roland et al. noted higher fasting glucose and impaired glucose tolerance in prenatally androgenised female mice which closely mimics PCOS model. 37 Prior to this, Nilsson et al. showed that exposure to testosterone in the neonatal period resulted in insulin resistance, change in adipose tissue distribution and lean mass without significant changes in circulating androgen concentrations. 102 Further, their groups showed this early exposure to androgen resulted in long-lasting effects on insulin sensitivity, adipose tissue and lipid profile. 103 We have previously published a detailed review of the abnormalities in adipose tissue function, fat distribution and lipid metabolism in the context of androgen excess. 104
We have demonstrated that expression of the androgen-activating enzyme, AKR1C3, which converts the weak androgen precursor androstenedione into potent testosterone, is increased in subcutaneous (SC) adipose tissue in women with PCOS. Locally generated androgens enhance de novo lipogenesis within SC female adipocytes, potentially predisposing to fatty acid overspill into the systemic circulation, with consequent lipotoxicity driving an adverse metabolic phenotype. 64 We demonstrated that associated insulin resistance and hyperinsulinemia then drive AKR1C3 expression and activity within the adipocyte, raising the possibility of a vicious circle of intra-adipose androgen activation, lipid accumulation and hyperinsulinaemia.
AKR1C3 also converts the C11-oxy C19 androgen precursor 11-ketoandrostenedione (11KA4) into 11-ketotestosterone (11KT), which binds and activates the AR with similar potency to testerosterone. 105 Recent data from Storbeck’s group also suggest that 11KA4 is the preferred substrate for AKR1C3, which more efficiently converts it to active 11KT than its counterpart reaction in the classic pathway with KM values of 0.11 μM and 0.06 μM for 11KA4 and A4 respectively. 106 Our findings of increased AKR1C3 expression in SAT of women with PCOS were corroborated by a recent study describing similar results. 107 Amer et al. have shown an upregulated AKR1C3 and CYP17A1 mRNA expression in SAT of women with PCOS as well as a higher levels of T. But, it is important to note that the authors did not assess if the higher levels of testerosterone in the adipose tissue was a result of increased AKR1C3 or CYP17A1 expression. This was previously shown by our group showing a reduction of testerosterone levels following inhibition of AKR1C3 by 3-4 trifluoromethyl-phenylamino-benzoic acid in healthy female adipose tissue. Studies on women with PCOS will be needed to confirm if the increased levels of testerosterone in SAT are in fact related to overexpression of AKR1C3 per se or related to both CYP17A1 and AKR1C3 activities. These findings define a novel intra-adipose mechanism that could contribute to androgen excess and metabolic dysfunction in PCOS.
These data strongly implicate adipose tissue as a major peripheral site of activation and metabolism of C11-oxy C19 androgens in PCOS and suggest that locally generated C11-oxy C19 androgens play a significant role in adipose tissue lipotoxicity. To date, however, this hypothesis has not been tested in adipose tissue of human participants using in vivo physiology approaches.
The association between adiposity, androgen excess and PCOS has been further complicated by hyperinsulinaemia and insulin resistance, which exacerbate the metabolic, reproductive and steroidogenic abnormalities observed in the disorder. 8 A systematic review by Lim et al. 96 found women with obesity and PCOS have a higher total and free testerosterone and lower SHBG compared with those with normal BMI. Furthermore, women with both PCOS and obesity also have a more severe clinical evidence of hyperandrogenism (such as hirsutism, menstrual abnormalities and anovulation) compared with women with PCOS and normal BMI. This effect tends to be more pronounced in the abdominal/visceral obesity phenotypes96,108–110 and weight loss in women with PCOS has been shown to improve clinical, metabolic and reproductive outcomes.110–112
Insulin resistance and compensatory hyperinsulinemia in obesity inhibits hepatic production of SHBG, with a consequent increased bioavailability of androgens. Further, insulin modulates hypothalamic-gonadal and steroid enzymatic machinery at various levels as described previously.
Pancreas, type 2 diabetes and androgen excess
Studies have shown that there is a clear sex difference in the way the foetal pancreas reacts to androgen excess. Insulin secreted from the pancreatic beta cells is critical for glucose utilisation in the peripheral tissues. Defects in insulin secretion or action predispose to insulin resistance and T2DM. Beta cells expand rapidly during late gestation and any alteration to its expansion during this period results in long-lasting deleterious effects. Navarro et al. demonstrated potentiating effects of DHT on male mice beta cell function via the glucagon-like peptide-1 (GLP-1) receptor. 113 Further, they showed male mice lacking AR had decreased glucose-stimulated insulin secretion, leading to glucose intolerance. Castration of male rats resulted in approximately 30% reduction in beta cell mass. 114 Xu et al. studied the transcriptome of AR-deficient islet beta cells of male mice and found altered expression of genes involved in inflammation and insulin secretion. 115
Studies in women with PCOS have also shown that androgen excess is proportionally related to beta cell dysfunction. This has been discussed in detailed by Mauvaus-Jarvis in a review of sex steroid effects on pancreatic beta cell function. 116 This hypothesis was further tested in female mice by exposing them to chronic androgen excess, which resulted hyperinsulinemia and insulin resistance. However, this was not observed in female βARKO mice lacking AR expression in pancreatic beta cells. 117 Testosterone prevents beta cell apoptosis and increases insulin mRNA levels both in vitro and in vivo in a series of studies conducted by Morimoto et al.118,119
Harada et al. demonstrated an in vivo effect of androgens on beta cell mass and expansion in a sex-specific manner. They found reduced beta cell mass and proliferation in male rats following administration of flutamide to pregnant dams. Although beta cell mass was restored after feeding the mice a high fat diet, they had persisting glucose intolerance suggesting decreased insulin secretion. 120
Androgen exposure during intrauterine life results in several phenotypic characteristics of PCOS in nonhuman primates and sheep. When rats were exposed to testosterone during late gestation, their female offspring exhibited significant weight gain, increased adipose tissue and other traits of metabolic dysfunction. 121 Exposing rhesus monkey dams to testosterone pre-conception resulted in hyperinsulinaemia and a reduction in glucose clearance following accelerated weight gain during testosterone treatment. This transient hyperglycaemic and relative hyperinsulinaemic episodes were sufficient to induce differential programming of insulin action and secretion in their female offspring. 122 Intrauterine exposure to testosterone resulted in increased beta cell numbers in prenatally androgenised female foetuses with no effect on alpha cells. 123 The same group reported upregulation of genes involved in beta cell development and function. 124 Neither studies found any such changes in male offspring, even when they were exposed to androgens before the male programming window. Roland et al. reported fasting hyperglycaemia and impaired glucose tolerance independently of age and changes in body composition or peripheral insulin sensitivity in prenatally androgenised female mice. Once again, no such changes were noted in male counterparts. 37
Liu et al. 125 demonstrated improved differentiation of pluripotent stem cells into insulin-producing cells when testosterone was added to routine differentiation medium, resulting in an upregulation of NGN3, NEUROD1 and INS genes. Zhang et al. investigated the association between free-androgen index, a marker of androgen excess, with glucose tolerance in PCOS. They found a positive correlation suggestive of beta cell dysfunction in women with PCOS. 126
Glucose-stimulated insulin secretion significantly decreases in islets treated with DHT in mice studies. This is associated with significant reductions in expression of several key genes involved in islet cell mitochondrial biogenesis and mitochondrial ATP production. 127 Chronic exposure to androgens resulted in increased oxidative stress in islet cells resulting in secondary beta cell failure in female mice fed on a western diet.117,128 The study also reported direct androgen receptor-dependant impairment of beta cell function by inducing mitochondrial dysfunction in vitro. 128 This is important in connection to skeletal muscle insulin resistance and failing insulin secretion would predispose women with PCOS to develop T2DM.
There are limitations that prevent direct translation findings from in vitro and animal studies into human physiology. Some of these limitations may be due to variability in animals for study, methods of randomization, choice of comparison therapy, blinding investigators to interventions and analysis, and variable duration of follow up.129,130 Nevertheless, these studies provide plausible pathophysiology and mechanisms linking pancreas and androgen excess, which have been explored in a few human studies described in the following.
PCOS is strongly linked with insulin resistance and T2DM in population and other large-scale studies. A meta-analysis of 35 studies of women with PCOS by Moran et al. reported increased odds for insulin resistance (OR 2.54, CI 1.44–4.47) and T2DM (OR 4.00, CI 1.97–8.10) compared with BMI-matched studies. 131 Legro et al. conducted one of the earliest prospective studies to assess the prevalence of T2DM in PCOS. They found the prevalence of T2DM in women with PCOS to be 7.5%; it was 1.5% in lean women with PCOS, further supporting the synergistic harmful effect of obesity on insulin sensitivity in PCOS. Interestingly, 31.1% of their cohort had impaired glucose tolerance speculating whether standards of diagnosing T2DM may not identify all index cases in PCOS cohort. 132 Another study by Glintborg et al. later found no differences in the prevalence of Impaired Glucose Tolerance (IGT) and T2DM in healthy weight women with PCOS compared with healthy controls. 133 Boudreaux et al. reported an incidence of 13.4% and 5.8% in women with PCOS and healthy controls respectively, with a relative risk of 2.3-times after prospective follow up over 8 years. Further, obesity increased the risk by fivefold in their cohort. 134 One of the largest population-based survey of T2DM in PCOS was reported from Australia by Joham et al. They surveyed 9145 women who self-reported the diagnosis of PCOS, T2DM and gestational diabetes mellitus. The prevalence of T2DM was 5.1%. After adjusting to sociodemographic and other known risk factors, the odds for T2DM were significantly increased in PCOS [odds ratio 8.8, 95% confidence interval (CI) 3.9–20.1, p = 0.001]. BMI was independently associated with T2DM in PCOS with every BMI increment increasing the risk of T2DM by 10%. 135 A Danish national register study by Rubin et al. studying about 18,000 women with PCOS and 54,000 controls has also shown a higher event rate of T2DM in PCOS compared with well-matched controls, and diabetes was diagnosed on average 4 years earlier than in the background population. 136 The prevalence of glucose intolerance and T2DM in a cohort of 122 women with PCOS studied by Ehrmann and colleagues was 35% and 10% respectively. 137 They further followed up a subset of the glucose-intolerant women with PCOS and found them to have higher post-prandial glucose compared with their baseline, suggesting a progressively worsening insulin resistance in the absence of any intervention. The prevalence of diabetes in the Dutch population was much less (2.3%) compared with the American cohorts described previously. However, the risk was 2.3-times higher compared with the generally healthy cohorts. And this risk increased from 1.3-times in 25–34 years to 9.4-times in 45–54 years. 138 The study by Kauffman and colleagues suggests that ethnicity plays an additive effect on insulin resistance in PCOS. Mexican American women had significantly higher insulin resistance compared with white American women, thus challenging a single population-wide screening tool. 139
First reported in 1980, the association of hyperandrogenism and hyperinsulinemia is firmly established.64,140–142 Our group have also shown in a retrospective cohort study in a UK primary care database that women with increased serum testosterone levels have an increased risk of incident T2DM. 143 This was recently highlighted further by Ruth et al. in their study of 455,097 UK Biobank samples, which showed that the risk of T2DM in women was increased by 37% for every increased standard deviation of free testosterone from baseline. A higher fasting insulin was also reported in these women. 144 However it is important to note that these are associations rather than causation and hence more prospective studies will be needed to investigate the impact of androgen excess on the development of T2DM.
Dunaif et al. were amongst the first to prove that women with PCOS have significant insulin resistance independent of obesity. However, obesity seemed to have a synergistic deleterious effect on glucose tolerance. 142 Further, they proved in their study that insulin resistance was not due to decreased insulin clearance but of a different pathology. 145 Since then, several researchers have studied the exact mechanisms of insulin resistance and its consequent deleterious effect on metabolism. Hypotheses to explain intrinsic insulin resistance in PCOS include mitochondrial dysfunction and lipid accumulation affecting the insulin-signalling pathway. Hansen et al. 146 found that lean women with PCOS have 25% lower insulin sensitivity and 40% lower plasma adiponectin levels compared with age- and BMI-matched controls. The finding of low levels of adiponectin has been shown to predict women with PCOS who are at high risk for developing T2DM. 147 They also reported an increased accumulation of triacylglycerol, diacylglycerol and ceramide in skeletal muscles of PCOS women supporting the lipid accumulation theory.
Muscle
Androgen excess may play a role impacting skeletal muscles in women with PCOS by altering their insulin sensitivity. Muscle is one of the key organs responsible for disposing 70–80% of glucose load. Insulin stimulates a canonical signalling cascade composed of insulin receptor substrate (IRS)-1, phosphoinositide 3-kinase (PI3K), protein kinases PDK1, Akt and Rab. The overall cascade ends with translocation of glucose transporter (GLUT) 4 to the cell membrane which permits the intake of glucose from blood into the cell. 148 Several groups have tried to tease out the role of androgen excess in metabolic dysfunction using hyperglycaemic and euglycaemic-hyperinsulinaemic clamps. Most of these studies concluded that high levels of testosterone resulted in reduction in whole-body glucose uptake in healthy women.149–152 Furthermore, they reported that these testosterone-induced insulin resistance were not attributed to hepatic insulin resistance supporting the role of muscles in androgen-mediated insulin resistance. 153 Insulin resistance is a common finding in PCOS. Stepto et al. found that 75% of lean women with PCOS and 95% of overweight women with PCOS are intrinsically insulin resistant. 154 Various scientists have attempted to explain insulin resistance in PCOS by one of three mechanisms: (1) dysregulation and/or dysfunction at several steps in the insulin-signalling cascade, (2) lipid accumulation and (3) mitochondrial dysfunction.155,156
Most of the theories about a dysfunctional insulin-signalling pathway come from studies on diabetes. The earliest proposed mechanism of insulin resistance in skeletal muscle of women with PCOS was increased phosphorylation of serine residue on IRS-1 limiting the signal cascade. 157 This was also shown in a later study by Corbould et al. 158 However, these findings could not be replicated by other groups. Instead, defects distal to IRS-1/IRS-2 involving Akt substrate 160 kDa159,160 and other pathways were found. This theory is supported by a study on the impact of exercise on hyperandrogenized mice, showing improvement in insulin sensitivity via PI3K-Akt pathway with associated reduction in 5αR1 expression in skeletal muscle of the exercise group versus stationary group. 161 A cross-sectional study looking at impact of habitual physical activity on women with PCOS showed an association of having more than 7500 steps per day (active group) with reduction of BMI, waist circumference, lipid accumulation product, androgen levels and fasting and 120-min insulin levels. In their study, HOMA-IR and 2000 daily steps increment were also found to be an independent predictor of free-androgen index. 162 A randomized trial on women with PCOS have also shown improvement in some androgen levels (total testosterone and SHBG) as well as insulin resistance shown by the decrease in HOMA-IR following 12 weeks of high-intensity interval training, further strengthening the link between androgens excess and insulin resistance in skeletal muscle. 163
Nilsson et al. found aberrant gene expression and DNA methylation in skeletal muscles of women with PCOS, mainly DYRK1A, which encodes an inhibitor of glycogen synthase kinase-3, SCP2, which is involved in lipid metabolism, SYNPO2, which encodes synaptopodin protein involved in oxidation in the muscles, KLF10, which is a transcriptional repressor regulating circadian expression of various genes involved in lipid and glucose metabolism, and NAMPT, which encodes visfatin and promotes glucose uptake into skeletal muscle. 164 However, many of these changes had contrasting results in the presence of androgens in vitro and hence remain inconclusive.
The other commonly proposed mechanisms explaining insulin resistance in PCOS include lipid accumulation and mitochondrial dysfunction. Lipid accumulation in skeletal muscle influencing glucose-regulating pathways is hypothesised to cause insulin resistance in diabetes.165,166 Intramuscular lipid levels (triacylglycerol, sn-1.3 diacylglycerol and ceramide) were higher in women with PCOS compared with healthy controls in a study done by Hanssen et al. In this study, they found women with PCOS had 25% higher whole-body insulin resistance compared with healthy controls. They also found lower AMP-activated protein kinase and Thr172 phosphorylation in association with lower plasma adiponectin levels suggesting a role of the latter in insulin resistance. 146 Studies on the theory of mitochondrial dysfunction causing insulin resistance has resulted in contrasting evidence over the last decades.167,168 A nontargeted metabolomics analysis of skeletal muscle of mice which was treated with DHEA revealed 32 metabolites and five metabolic pathways that are significantly different compared with controls. 169 Among these, the reduced NAD+/NADH ratio affecting ATP generation was a key finding, which may influence downstream activation of the insulin-signalling pathway, supporting the mitochondrial dysfunction theory. Skov et al. showed that impaired insulin-stimulated glucose disposal in women with PCOS was associated with downregulation of mitochondrial oxidative phosphorylation genes using global genetic pathway analysis. 170 Interestingly, Hutchinson et al. could not explain the difference in insulin resistance between women with and without PCOS with either lipid accumulation or mitochondrial dysfunction. 171 The ability of high-intensity interval training to improve insulin sensitivity and androgen excess may also be related to increase in mitochondrial density and capacity during the training as well as increased in fat oxidation reducing lipid accumulation.163,172
Taken together, the essence of these studies suggests there is perhaps a distinct mechanism of insulin resistance in PCOS. Future studies on the specific influence of androgens on insulin resistance in skeletal muscle may answer this question.
Liver
Several cross-sectional studies have shown an increased prevalence of NAFLD in women with PCOS; NAFLD is now on its way to become the most frequent cause of liver transplantation, driven by the global obesity pandemic, and is a forerunner of CVD. 173 A study by Petta et al. concluded that PCOS is an independent risk factor for hepatic steatosis and possibly progression to fibrosis and cirrhosis, with hyperandrogenism and insulin resistance as main determinants. 174 These associations have been observed in other studies.175–177 Our group has carried out a cohort study utilising a large primary care database in the United Kingdom, including 63,000 women with PCOS and 121,000 matched controls, revealing that the risk of NAFLD was significantly increased in women with PCOS, even in women with a normal BMI (hazard ratio = 2.23, 95% CI 1.86–2.66, p < 0.001). 178 Androgen excess (high testosterone, low SHBG) was found to be a contributing risk factor for the development of NAFLD in PCOS in this study. 178 These associations have been recently supported by a meta-analysis showing a 2.3-fold increased rate of NAFLD in women with PCOS, especially those who have hyperandrogenism. 179
There are numerous proposed mechanisms linking this hepatic manifestation of metabolic syndrome with PCOS.179,180 Androgen excess in PCOS suppressed low-density lipoprotein receptor RNA expression both in the adipocytes and the liver.They speculate the suppressed receptor expression might prolong plasma half-life of Very low density lipoprotein (VLDL) and low density lipoprotein (LDL), potentially leading to lipid accumulation both in the adipocytes and liver. 181 Tumour necrosis factor (TNF)-α levels related to hyperandrogenism have also been implicated in the development of NAFLD. 182 DHEA-induced hyperandrogenism in healthy women of reproductive age resulted in increased fasting AR mRNA content and TNF-α levels, which were both potentiated by glucose ingestion. 183 TNF-α is one of the proinflammatory cytokines involved in many inflammatory disorders, including metabolic syndrome and has also been shown to induce insulin resistance via promotion of IRS-1. 184 TNF-α is also involved in inducing enzymes involved in lipid metabolism, proinflammatory cytokines and fibrosis-associated protein in the liver, thus playing a pivotal role in development of NAFLD. A study by Shimomura et al. showed that the adipocyte-specific nuclear form of sterol regulatory element-binding protein 1c (nSREBP-1c) transgenic mice developed hepatic changes similar to the ones seen in NAFLD, with increased TNF-α. 185 Follow-up studies by Kakino et al. using the same mouse model showed that TNF-α is responsible for the development of NAFLD. 182 In this study, TNF knockout nSREBP-1c mice showed an improved glucose tolerance and there was a significantly reduced prevalence of hepatic steatosis compared with the original nSREBP-1c model. This finding was also supported by culturing primary hepatocytes in the presence of TNF-α. 182 These observations support that increased TNF-α, possibly mediated by hyperandrogenism, is an important mechanism in the development and progression of NAFLD in women with PCOS.
Our group has demonstrated androgen-mediated suppression of lipolysis and increased de novo lipogenesis in vivo and in vitro. 186 This would result in net positive fat accumulation beyond the adipocyte storing capacities causing fatty acid overspill, systemic lipotoxicity, insulin resistance and fat accumulation in the liver, thus leading to the development of NAFLD in women with PCOS. In the same study, serum metabolomics showed increased concentration of glycerophospholipids and lysoglycerophospholipids in women with PCOS with androgen excess, but not in controls with normal androgen concentrations, at baseline. Acute androgen exposure yielded a further increased in these metabolites whereas a decrease was observed in the BMI-matched healthy controls. 64 Both glycerophospholipids and lysoglycerophospholipids were previously observed to be increased in people with NAFLD and have been identified as potential markers of risk and progression of the condition. 187 These data shows that women with PCOS have a distinct metabolic response to androgens which might contribute to the development of NAFLD and other related conditions.
The role of microRNA
Interestingly, miRNA differential expression was previously associated with the increased risk in the development of T2DM.188,189 These expressions have been investigated in women with PCOS in the context of insulin resistance (IR). 71 miR-222 has been shown to be positively associated with hyperinsulinaemia in women with PCOS, suggesting its role in IR in PCOS. 69 Treating an IR adipose tissue cell line with high levels of glucose and insulin increased levels of miR-320 significantly which in turns reversed the IR via increasing the expression of GLUT4. Moreover, miR-320 have been found in the follicular fluid of women with PCOS which may present a future therapeutic target for women with PCOS and IR. 190 Jiang et al. added to the literature of miRNA and IR in PCOS later showing the upregulation of miR-122, miR-193b and miR-194 in women with IGT and PCOS compared with those with normal glucose tolerance. miR-33b-5p expression in ovarian tissues has also been found to play a role in propagating IR in PCOS. 191 This miRNA has been found to be increased in rats with PCOS and IR with levels negatively correlated with the expression GLUT4, high mobility group A2 (HMGA2) and sterol regulatory element-binding protein 1 (SREBF1). The overexpression was also shown in IR adipose tissue in vivo with a similar reduced expression of GLUT4, HMGA2 and SREBF1. Furthermore, expression of GLUT4, HMGA2 and SREBF1 was increased following the inhibition of miR-33b-5p, further strengthening the role of this miRNA in promoting IR in PCOS via this pathway.
The role of miRNA, obesity and dyslipidaemia have also been discussed by Chen et al. in their review. 71 Several studies have also shown differential expression of miRNA with their expressions correlating to markers of adiposity such as BMI and waist-to-hip ratio.72,78,192 Arancio et al. have also shown association of miRNA levels with LDL cholesterol levels in women with PCOS and hyperandrogenism, adding into the evidence on androgen excess and miRNA in propogating metabolic diseases in women with PCOS. 73
In summary, miRNAs have been shown to be play a role in the metabolic derrangements in women with PCOS with evidences linking them to androgen excess. More studies will be needed to thoroughly investigate these associations to enable for successful therapeutic targets specifically for reducing the metabolic risk in women with PCOS.
Androgen excess and cardiovascular disease
Earlier studies investigating the association between PCOS and CVD reported no increased prevalence. Pierpoint et al. reviewed the case notes of 786 women who were diagnosed with PCOS between 1930 and 1979 and were followed up for an average 30 years. They did not find any increased deaths due to CVDs in this cohort compared with national rates. 193 Interestingly, the same group reported higher prevalence of cardiovascular risk factors and nonfatal cerebrovascular disease in this cohort. 194 While they concluded that PCOS may have protective effects on CVD, most of these patients did not have a hormonal profile, challenging the diagnosis and the conclusion of the study. Dahlgren et al. reported a 7.4-times higher risk for myocardial infarction in women with PCOS compared with those without, using an early risk factor model. 195 However, the number of participants was relatively small. Birdsall et al. reported a more extensive coronary artery disease in postmenopausal women with polycystic ovaries on ultrasound. 196 Although the the diagnosis of PCOS in this cohort could not be ascertained from their report, the study reported associations between the polycystic ovaries with hirsutism and high levels of free testosterone. Christian et al. found that women with PCOS had higher coronary artery calcification compared with their age-matched controls, independently of obesity. 197 The incidence of coronary artery disease was four-times higher in Czech women with a history of PCOS compared with the general population. 198 Although these women also had higher prevalence of T2DM, the remaining cardiovascular risk factors were comparable with the rest of the population, questioning whether PCOS is an independent risk factor for coronary artery disease. 198 Based on these findings, the Androgen Excess and PCOS Society acknowledged that women with PCOS have a moderate-to-high risk for CVD, depending on the presence of other risk factors. 199
A meta-analysis evaluating the effect of androgen excess in PCOS on metabolic parameters has shown that androgen excess is associated with higher levels of total cholesterol and lower high-density cholesterol levels. 200 A study by Luque-Ramírez et al. comparing hyperandrogenic with nonhyperandrogenic PCOS showed an increased mean carotid intima-media thickness independently of BMI, with the main determinant being the concentration of serum total testosterone and A4. 201 Furthermore, testosterone has also been shown to inhibit bradykinin-induced intracellular calcium kinetics resulting in endothelial dysfunction. 202 Androgens are also known to impact the renal system through the upregulation of sodium channels in the proximal tubules increasing the rate of fluid reabsorption, hence increasing extracellular volume and blood pressure.203,204 Orio et al. 205 clinically confirmed endothelial dysfunction in brachial arteries and increased cardiac intima-media thickness directly proportional to androgen excess and independently of obesity in PCOS compared with age- and BMI-matched controls. Kravariti et al. and Luque-Ramírez et al. independently confirmed both these findings in PCOS women of differing ethnicities.201,206 Levels of serum highly sensitive C-reactive protein (hsCRP) has been previously shown to be higher in women with PCOS, which may play a role in increased risk of CVD in this population.207,208 Möhlig et al. later found that increased levels of hsCRP were due to obesity rather than PCOS alone 209 , which was confirmed by another study showing increased hsCRP in women with PCOS that diminishes after adjustment for BMI. The study also reported no association between levels of androgens and that of hsCRP. 210 Hyperhomocysteinaemia and oxidative stress are widely known as risk factors for the development of CVD.211,212 Several studies have previously shown elevated levels of homocysteine and oxidative stress in women with PCOS which may contribute to their risk of developing CVD.213–216 A study by Yilmaz et al. 217 also showed a positive correlation between levels of oxidative stress and free testosterone levels in women with PCOS, suggesting the potential role of androgen excess in increasing risk of CVD via this mechnanism. Although there is no evidence on the relationship between androgen excess and homocysteine levels, many studies using anti-androgenic oral contraceptives containing 35 μg ehtynylestroadiol and 2 mg cyproterone acetate have demonstrated rapid reductions of homocysteine levels in women with PCOS.218–220 However, this was not the case for anti-androgen-containing drosperinone. 221 Recently, the role of androgens in attenuating endothelin-1-induced vasodilation and endothelin B receptor-mediated nitric oxide production resulting in endothelial dysfunction was described by Usselman et al. in women with PCOS. 222 These and other studies support that androgen excess is implicated either directly or indirectly in almost all proposed mechanisms to explain the increased risk for CVD in PCOS.
Conclusion
Androgen excess plays a pivotal role in increasing risk of metabolic dysfunction in women with PCOS. Currently, there are no disease-specific therapeutic options available to modify metabolic risk in women with PCOS, and treatments are limited to the use of insulin-sensitising agents, anti-hypertensives and lipid lowering agents. Consensus guidelines 2 have consistently acknowledged the need to identify novel biomarkers and therapies for metabolic risk in women with PCOS, and targeting androgen excess is likely to represent the most promising future therapeutic avenue. With the emerging role of the less-well-characterised C11-oxy C19 androgen subclass, it is important to establish the origins and roles of specific androgens in metabolic pathophysiology in order to identify potential therapeutic targets. There is now a strong rationale for therapies targeting androgen synthesis or action for the amelioration of metabolic risk in women with PCOS.
Footnotes
Author contribution(s)
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
This review paper is supported by WA’s Wellcome Trust Investigator award grant and Emerging Clinician Scientist Award (HRB ECSA-2020-001) to MOR.