
Abstract
Acquired von Willebrand syndrome (AVWS) is a well-known complication of a monoclonal gammopathy with a potentially severe bleeding tendency. Treatment with von Willebrand factor (VWF)/factor VIII (FVIII) concentrate yields mixed results in controlling the bleeding diathesis, while the use of intravenous immunoglobulins may be effective. However, clear guidelines for the optimal management of AVWS are lacking. Therefore, we retrospectively analyzed the cases of AVWS secondary to monoclonal gammopathy at the University Hospitals of Leuven. We confirm the beneficial effect of intravenous immunoglobulins in IgG-associated AVWS. For IgM-associated AVWS, we observed better results with the administration of VWF/FVIII concentrate or a combination of therapies. Of note, one patient with IgG-associated AVWS did not respond to immunoglobulins and had mutations in the VWF and fibrinogen gamma chain (FGG) genes. This report adds additional cases to the literature of this rare cause of acquired bleeding.
Keywords
Introduction
Acquired von Willebrand syndrome (AVWS) is a rare bleeding disorder with a bleeding phenotype comparable to congenital von Willebrand disease (VWD). Both entities can be distinguished by the absence of a personal and family history of bleeding diathesis in AVWS. 1 It is mainly observed in association with cardiovascular disease and hematologic malignancies. Cardiovascular diseases associated with AVWS include aortic valve stenosis, also known as Heyde’s syndrome when associated with gastrointestinal bleeding, 2 hypertrophic cardiomyopathy, extracorporeal membrane oxygenation, and left ventricular assist devices. Hematologic malignancies include lymphoproliferative diseases, including plasma cell dyscrasias, and on the other hand myeloproliferative neoplasms, such as essential thrombocythemia. Other underlying conditions include autoimmune diseases, hypothyroidism, solid organ tumors, and certain drugs. 3 Typically, AVWS due to lymphoproliferative disease leads to more severe bleeding complications than AVWS associated with cardiovascular disease. 4
In this case series, we focused on AVWS secondary to monoclonal gammopathy (AVWS-MG), which is an important association in adults. AVWS is one of the conditions that complicates monoclonal gammopathy of unknown significance (MGUS), leading to the concept of monoclonal gammopathy of clinical significance (MGCS).
5
The bleeding symptoms of AVWS-MG can be treated in a similar manner to hereditary VWD with 1-deamino-8-
Current evidence on AVWS-MG comes from reported case series and an international registry3,9 in the absence of randomized controlled trials. Therefore, there are no strict recommendations for the optimal management of AVWS-MG. Therefore, we review our experience over the past 20 years regarding the characteristics and management of AVWS secondary to MG.
Methods
We retrospectively analyzed the data of adult patients who were diagnosed at University Hospitals Leuven, Belgium, between 2002 and 2023. In the report, patients are referred to as patients 1–7 in chronological order of diagnosis. All laboratory analyses were performed according to routine clinical practice. Von Willebrand factor antigen (VWF:Ag) and von Willebrand factor ristocetin cofactor (VWF:RCo) levels were measured by chemiluminescent assays using an AcuStar according to the manufacturer’s instructions. 10 Before April 2014, the samples were analysed on a BCS/BCS XP coagulation analyser (Siemens; Marburg, Germany) with the VWF:Ag and BC von Willebrand Reagent (VWF:RCo) kits (Siemens), according to the manufacturer’s recommendations. Factor VIII (FVIII) one-stage assays were performed on an ACL-TOP700 (Werfen), with the manufacturer’s standard protocol and reagents (HemosIL SynthASil). A complete response to a treatment was defined as attaining VWF:Ag levels above 50%, a partial response was defined as attaining VWF:Ag levels between 20% and 50%. Sequencing was done on NovaSeq 6000 (Illumina) using the Twist Biosciences Exome CustomV2 panel. 11 Continuous variables are expressed as median and interquartile range (IQR), and categorical variables are expressed as a number (n). Graphs were generated using GraphPad Prism (version 10.1.1), Dotmatics.
Results
Seven patients were diagnosed with AVWS-MG (Table 1). Five of the seven patients were male. The median age at diagnosis was 60 years (IQR: 41.5–64 years). Six out of seven patients received their diagnosis of MGUS according to the International Myeloma Working Group 12 after the diagnosis of the AVWS (n = 6) with either monoclonal IgG (n = 5) or IgM (n = 1). One patient was diagnosed with Waldenström’s macroglobulinemia prior to the diagnosis of AVWS (patient 7). During follow-up, patient 2 progressed to smoldering myeloma and patient 1 to multiple myeloma (see Supplemental Table 1). None of the patients had a personal or family history of spontaneous bleeding or increased bleeding after surgery. At diagnosis, patients presented with spontaneous cutaneous hematoma (n = 3), spontaneous mucosal bleeding (n = 2), increased or prolonged bleeding after surgery (n = 4), gastrointestinal bleeding (n = 1), and/or large post-traumatic hematoma (n = 1). Two patients had a decrease in hemoglobin of 2 g/dl or more, and required red blood cell transfusion at the time of diagnosis, and these bleeds were therefore classified as major bleeds according to International Society on Thrombosis and Hemostasis criteria. 13 In six of the seven patients, the bleeding was the initial symptom, and the underlying MG was diagnosed only during subsequent workup. This was in contrast to patient 7, who had been diagnosed with Waldenström’s macroglobulinemia prior to the acquisition of an increased bleeding tendency.
Patient characteristics and lab values at diagnosis.
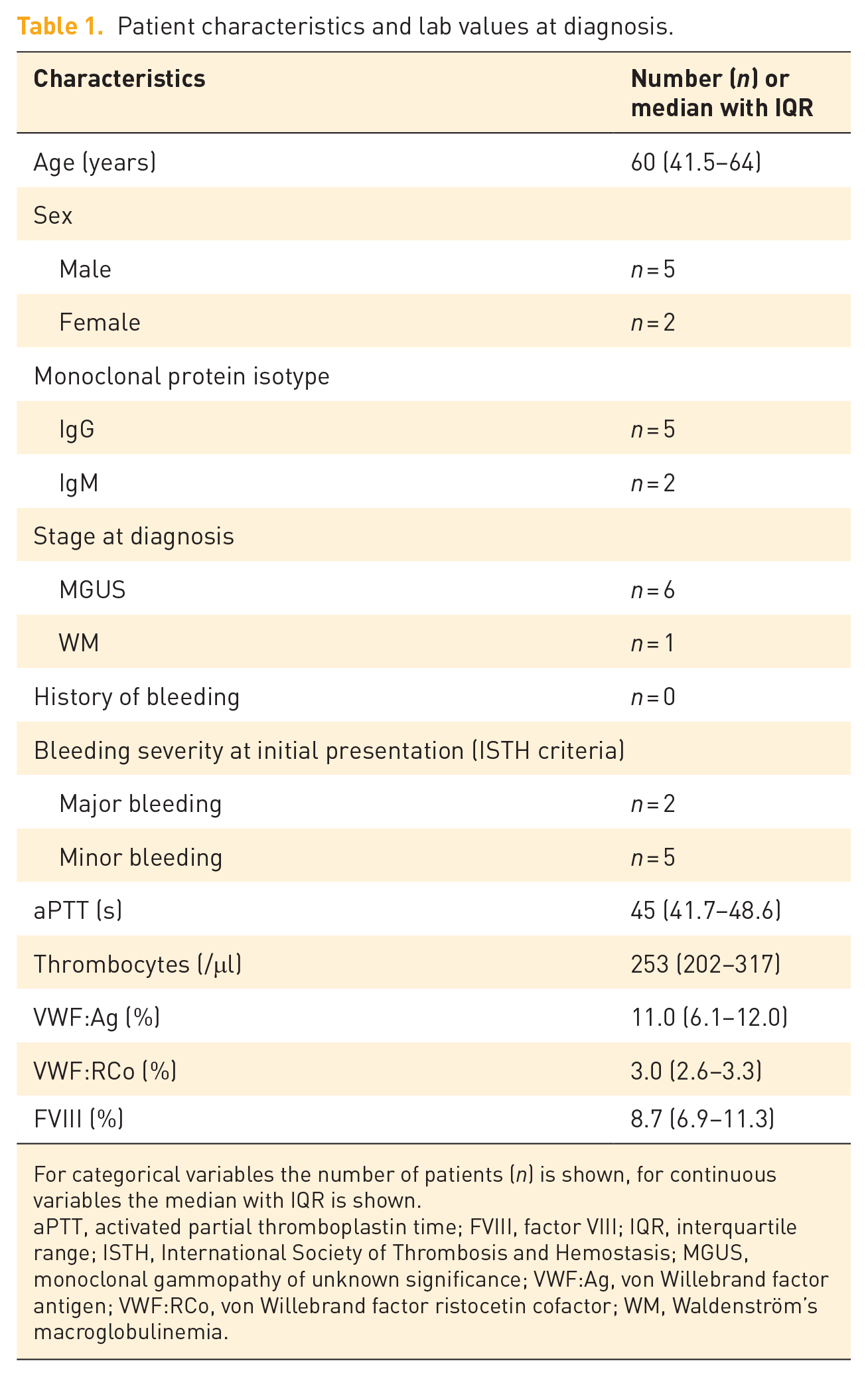
For categorical variables the number of patients (n) is shown, for continuous variables the median with IQR is shown.
aPTT, activated partial thromboplastin time; FVIII, factor VIII; IQR, interquartile range; ISTH, International Society of Thrombosis and Hemostasis; MGUS, monoclonal gammopathy of unknown significance; VWF:Ag, von Willebrand factor antigen; VWF:RCo, von Willebrand factor ristocetin cofactor; WM, Waldenström’s macroglobulinemia.
Laboratory results at diagnosis showed that the VWF:RCo was decreased to a greater extent than VWF:Ag, indicating a VWD type 2 phenotype in the majority of cases (median VWF:RCo 3.0%, IQR 2.6–3.3; median VWF:Ag 11.0%, IQR 6.1–12.0). The VWF:RCo results for three patients were reported as below our laboratory’s threshold and were subsequently censored from further analysis. FVIII was also decreased (median 8.7%, IQR 6.9–11.3) with a subsequent increase in the activated partial thromboplastin time (aPTT; median 45 s, IQR 41.7–48.6). Full patient characteristics are provided in the supporting information (Supplemental Tables 1–4).
Our patients received various treatments either to treat the bleeding or to evaluate the effect on coagulation parameters to correct the bleeding diathesis: DDAVP, VWF/FVIII concentrate, and IVIGs. DDAVP was tried in three patients with IgG MGUS (doses 12–20 μg, see Supplemental Table 4), and resulted in a rapid, but short-lived response in only one patient (Figure 1). VWF/FVIII concentrate (Haemate P, CSL Behring; or Wilate, Octapharma, Belgium) was used in six patients (dose 33–50 IU/kg, see Supplemental Table 3). The effect is shown for four patients only (Figure 2), as the individual effect of factor replacement therapy on the VWF:Ag levels was not tested in the other two patients. Patient 2 (IgG) and patient 7 (IgM) showed a transient increase in VWF:Ag levels after administration of VWF/FVIII concentrate. However, the other two patients (patients 1 and 5) did not respond with VWF:Ag levels above 50%. IVIG (Privigen, CSL Behring) at 1 g/kg for 2 days was evaluated in all patients, either as a treatment for bleeding or to assess the effect on coagulation parameters. Only patient 7, with a known IgM gammopathy, did not receive IVIG. He had a complete response to VWF/FVIII concentrate. Three out of five patients with IgG MGUS had a complete response (VWF:Ag or VWF:RCo of at least 50%) to IVIG. Patient 5 had a partial response (reaching VWF:Ag levels between 20% and 50%) with VWF:Ag increasing from 8% to 42% at 24 h, and patient 1 had a minimal response with VWF:Ag increasing from 5.7% to 10.8% at 24 h (Figure 3). The duration of the effect was poorly documented, but the responses lasted from several days to weeks (see Supplemental Table 2). The remaining patient with IgM MGUS (patient 6) was treated with a combination of VWF/FVIII concentrate, rituximab (375 mg/m2) and methylprednisolone (32 mg) due to a lack of clinical benefit from IVIG. Finally, tranexamic acid was prescribed to all patients with minor bleeding, or around surgery in addition to the above measures. The different treatments and their respective effects are summarized in Table 2.
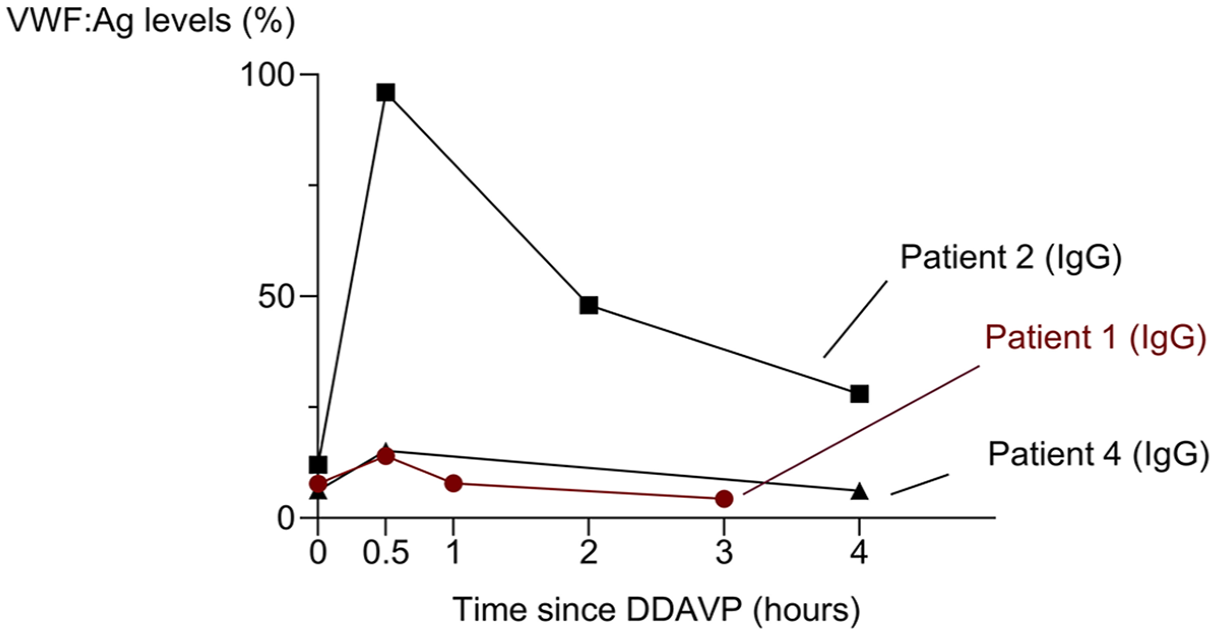
The VWF:Ag levels only increased in one patient out of three after DDAVP. The increase in VWF:Ag levels after administration of DDAVP is shown at the different time points for patients 1, 2, and 4. Patient 1 had a VWF mutation and is indicated in red.

The VWF:Ag levels increase in a subset of patients after administration of FVIII/VWF concentrate. The increase in VWF:Ag levels after administration of DDAVP is shown at the different time points for patients 1, 2, 5 and 7. One patient with IgM responded (patient 7) and one with IgG (patient 2). The other 2 patients did not reach levels above 50%. The patient with a VWF mutation (patient 1) is indicated in red.

The VWF:Ag levels (a) and VWF:RCo (b) increased after administration of IVIG in the majority of cases with IgG MGUS. The increase in von Willebrand factor antigen (VWF:Ag) and von Willebrand ristocetin cofactor (VWF:RCo) levels after administration of DDAVP are shown at the different time points for patients 1, 2, 3, 4, 5, and 6. Three out of five patients with IgG MGUS (patients 2–4) responded to IVIG (VWF:Ag and VWF:RCo > 50%). Out of the two other cases, one had an additional VWF mutation (patient 1, indicated in red). The patient with IgM MGUS (patient 6) did not respond.
The different treatments are shown with their effects on VWF:Ag levels.

“V” indicates a complete response (VWF:Ag > 50%), and “X” a partial (VWF:Ag 20%–50%) or no response (VWF < 20%). “O” indicates that a treatment was not administered or that the treatment response was not assessed. Other treatments include myeloma treatment (bortezomib-lenalidomide-dexamethasone induction, autologous stem cell transplantation, lenalidomide maintenance) for patient 1, rituximab and methylprednisolone for patient 6, zanubrutinib for patient 7. All patients received tranexamic acid.
DDAVP, 1-deamino-8
After the diagnosis of AVWS-MG was established, the subsequent treatment was selected based on the initial treatment results. Patient 1 received a combination of IVIG and VWF/FVIII concentrate before dental extraction with good hemostatic efficacy. Patient 2 received IVIG as monotherapy prior to several procedures (adnexectomy, hip arthroscopy, and dental surgery), with hemostasis achieved. Patient 3 received IVIG after a traumatic hemorrhage of the right ear, and before urgent osteosynthesis of a femur fracture. He also received prophylactic IVIG prior to a colonoscopy with biopsies. In all cases, hemostasis was achieved. He had a suspected rebleeding after an urgent endoscopic retrograde cholangiopancreaticography, for which IVIGs were repeated with good clinical effect. Patient 4 had no bleeding requiring treatment during follow-up. He received two courses of IVIG before an extended vacation abroad. Patient 5 had recurrent epistaxis during follow-up. Hemostasis was achieved each time with a combination of IVIG, VWF/FVIII concentrate, and tranexamic acid in addition to local measures. After a traumatic proximal radius fracture with secondary hematoma, IVIG and VWF/FVIII concentrate were given with good effect. The combination of IVIG and VWF/FVIII concentrate was also administered prior to excision of a melanoma without bleeding complications. Patient 6’s initial rectal bleeding required multiple transfusions, and necessitated a combination of VWF/FVIII concentrate, IVIG, rituximab, and methylprednisolone to control the bleeding. After discharge, he continued to receive daily prophylactic VWF/FVIII concentrate. After recurrence of rectal hemorrhoidal bleeding, the patient underwent hemorrhoidal plexus embolization. Subsequently, VWF/FVIII prophylaxis was tapered to three times per week. Due to a partial recovery of the patient’s VWF levels (VWF:Ag 24 IU/dl and VWF:RCo 22 IU/dl at last visit vs VWF:Ag 9 IU/dl and VWF:RCo 2 IU/dl at diagnosis), VWF/FVIII prophylaxis was eventually discontinued. In later follow-up, VWF/FVIII concentrate was administered once before cataract surgery with good hemostatic control. Patient 7 received VWF/FVIII concentrate and tranexamic acid prior to elective carpal tunnel release. There were no bleeding complications.
Notably, one patient who did not respond to IVIGs (patient 1), underwent next-generation sequencing using our panel for unexplained increased bleeding tendency to differentiate between an AVWS and a late diagnosis of congenital VWD or a combination. This revealed two heterozygous mutations in the VWF gene, c.4751A>G (p.Tyr1584Cys), and the fibrinogen gamma chain (FGG) gene, c.323C>G (p.Ala108Gly). Both are class 4 mutations (likely pathogenic) 14 and have been described previously.15,16 The VWF mutation is associated with VWD. In addition, a class 3 variant of unknown significance as defined by the American College of Medical Genetics, 14 was found in F7, c.208C>TT (p.Pro70Ser).
In addition to therapies targeting the hemostatic defect, two patients were treated for their underlying disease. Patient 1 had progressed to multiple myeloma after 20 years of follow-up, and was treated with the standard induction regimen of bortezomib, lenalidomide, and dexamethasone (VRd). This was followed by an autologous stem cell transplantation after high-dose melphalan (200 mg/m2). After induction therapy VWF:RCo levels recovered from 2.3% to 32.5%, and VWF:Ag from 7.8% to 43.6%, with a further recovery of VWF levels after autologous stem cell transplantation and subsequent lenalidomide maintenance (VWF:Rco 91.5%, VWF:Ag 98.6%). Patient 7 had relapsed Waldenström’s macroglobulinemia at diagnosis. Prior to the diagnosis, he had been treated with rituximab and bendamustine in 2017, and only developed AVWS at relapse in 2023. He was subsequently treated with the Bruton kinase inhibitor zanubrutinib. After 4 weeks of treatment, we observed an improvement in anemia, a decrease in IgM (28.6–15.1 g/l, reference range: 0.46–3.04 g/l), and a concomitant increase in VWF:Ag (30.9%–55.6%), VWF:RCo (22.1%–45.4%), and FVIII levels (41.6%–65.0%).
Finally, we demonstrated a causal relationship between the monoclonal protein and the associated AVWS in patient 6. A patient-specific enzyme-linked immunosorbent assay (ELISA) demonstrated the binding of the monoclonal IgM to VWF, and this binding was inversely correlated with the dilution of the patient’s plasma (Figure 4). In a follow-up sample after the start of treatment with rituximab, this correlation was lost. The VWF levels partially recovered (VWF:Ag 9 IU/dl and VWF:RCo 2 IU/dl at diagnosis; VWF:Ag 24 IU/dl and VWF:RCo 22 IU/dl at last visit).

IgM antibodies of patient 6 bind to VWF. The binding of VWF is shown as an optical density at different dilutions of the plasma samples. The binding is lost with progressive dilution of the patient’s plasma (measured by ELISA). Follow-up samples after treatment with rituximab are depicted in blue. The patient’s platelets and ELISA in control plasma are shown.
Discussion
The relationship between MG and AVWS is an important, but often overlooked association. In most patients, the MG was only discovered after the development of bleeding symptoms. Mechanistically, the paraprotein binds to VWF, resulting in increased VWF clearance through the reticuloendothelial system. 1 It has previously been suggested that IgG preferentially binds to the larger VWF multimers, 17 explaining the VWD type 2 phenotype which is observed in our cohort and in most cases in the literature. 18 Another proposed mechanism is the adsorption of VWF by clonal plasma cells expressing glycoprotein Ib. 19 In contrast to acquired hemophilia A, inhibitory VWF-binding antibodies are rarely detected. 20 However, we were able to demonstrate a relationship between binding capacity and plasma dilution using ELISA in one patient, indirectly demonstrating the binding of the monoclonal protein to VWF. In addition, the management of this subtype of AVWS can be particularly challenging. Treatments may target either the hemostatic defect, or the underlying disease.
Hemostatic strategies which are adopted from the treatment of congenital VWD, including DDAVP and VWF/FVIII concentrate, have shown mixed results in our cohort. In the only prospective study on this matter, DDAVP and VWF/FVIII concentrate improved VWF levels only transiently. However, in IgG-associated AVWS, high-dose IVIG induced a durable response. 7 In our cohort, most patients with IgG-associated AVWS also had a durable response to IVIG. These may work by blinding the reticuloendothelial system, capturing the circulating immune complexes or may involve an anti-idiotype effect. 1 A larger, retrospective analysis of 89 patients from various case reports and case series, showed a similarly favorable laboratory response to IVIG in 88.7%. 9 In contrast, IgM-associated AVWS patients had no response in the study by Federici et al., 7 as did the only IgM patient treated with IVIG in our cohort. Other retrospective studies have confirmed this discrepancy in the effect of IVIG in IgG patients versus IgM patients. Because of the heterogeneous response to the different treatments, it is useful to administer a test dose of DDAVP, VWF/FVIII concentrate, and high-dose IVIG upon diagnosis to evaluate their respective effects on the VWF:Ag, VWF:RCo, and FVIII levels. 21 This is even more true for the patients with IgM-associated AVWS. IVIGs do not seem to work in this subgroup and are therefore not recommended. In addition, these patients have unpredictable responses to DDAVP and VWF/FVIII concentrate. In summary, patients in our cohort were treated according to the guidelines of the international registry by Federici et al. 3 Patients received either IVIG as monotherapy (patients 2, 3, and 4), a combination of IVIG and VWF/FVIII concentrate (patients 1 and 5), or VWF/FVIII concentrate alone (patients 6 and 7) in case of bleeding or before procedures, based on the initial treatment outcomes.
In acute hemorrhage, the effect of IVIG is too slow and VWF/FVIII concentrate is the preferred treatment used in combination with IVIG, resulting in a higher recovery of VWF/FVIII and a better hemostatic outcome. 22 Dane et al. reported the successful use of continuous infusion of VWF concentrate in three patients who failed to respond to a bolus dose of VWF concentrate. 23 Recombinant activated F7 is an option for refractory bleeds. 24 In addition to the above treatments, tranexamic acid may be used for minor bleeding and in the perioperative management. 1 Finally, emicizumab has been reported to provide good hemostatic coverage in one case report. 25
Treatment of the underlying plasma cell dyscrasia should theoretically abolish the autoimmune response to VWF. In the event of progression to multiple myeloma or Waldenström’s macroglobulinemia, treatment should consist of standard therapy for either disease.26,27 In our cohort, two patients were treated for multiple myeloma and Waldenström’s macroglobulinemia. In the patient with Waldenström’s macroglobulinemia, zanubrutinib was chosen over ibrutinib because of a slightly lower bleeding incidence reported in the ASPEN trial. 28 Improvements in hemostatic parameters were seen after only 4 weeks of treatment. Treatments that target the underlying plasma cell clone have also been tried in MGUS. Corticosteroids, rituximab, bortezomib, lenalidomide, cyclophosphamide, melphalan, and daratumumab have been reported with variable success.29–31 In cases of life-threatening bleeding, anti-myeloma therapy is recommended in addition to hemostatic therapy to treat the underlying MGCS. 32
Interestingly, in our series, one patient with an IgG paraprotein and poor response to IVIG treatment had additional mutations in VWF, FGG, and F7. The p.Tyr1584Cys variant in VWF is common and was associated with VWF levels below 50% and at least one bleeding symptom in 93% of patients in a recent report. 33 However, our patient’s VWF levels fully recovered after achieving a complete response to myeloma treatment. Compared to healthy controls, a higher VWF clearance was observed in the presence of the p.Tyr1584Cys variant. However, it remains unclear whether this variant influences the response to IVIG. Therefore, results from genetic testing of the VWF gene should be interpreted with caution. Genetic testing should be considered in conjunction with the patient’s clinical history: the patient’s blood group, the family history, and the bleeding diathesis preceding the diagnosis of MGUS.
Our case series is limited mainly by its retrospective nature, with missing data and some patients treated with DDAVP, others only with IVIG or VWF/FVIII concentrate. A second limitation is the lack of a more detailed diagnostic workup in all patients (e.g., VWF propeptide analysis, VWF multimer analysis, and VWF gene sequencing results).
Conclusion
In conclusion, AVWS is an important disorder associated with MG. Classical hemostatic strategies with DDAVP and VWF/FVIII concentrate have unpredictable results in these patients. In contrast, responses to IVIG are often seen, especially in IgG-associated AVWS, and this was confirmed in our cohort. Our report adds more clinical data to the literature and highlights the need for clear recommendations based on prospective studies confirming the benefit of IVIG in AVWS secondary to MG.
Supplemental Material
sj-docx-1-tah-10.1177_20406207251347235 – Supplemental material for Acquired von Willebrand syndrome secondary to monoclonal gammopathy: a single-center case series
Supplemental material, sj-docx-1-tah-10.1177_20406207251347235 for Acquired von Willebrand syndrome secondary to monoclonal gammopathy: a single-center case series by Quentin Van Thillo, Finn Segers, Jan Brijs, Ulrike Douven, Radha Ramanan, Michel Delforge, Ann Janssens, Cédric Hermans, Johan De Bent, Marc Jacquemin, Thomas Vanassche and Peter Verhamme in Therapeutic Advances in Hematology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.