
Abstract
Scleroderma renal crisis is a serious complication that can develop in certain patients with systemic sclerosis. Some risks have been identified as potential triggers of scleroderma renal crisis, including the high-dose oral corticosteroids. Here, we present a patient who developed clinically severe systemic sclerosis and scleroderma renal crisis after exposure to oral corticosteroids and intravitreal vascular endothelial growth factor blockade with bevacizumab for cotton wool spots. The patient’s scleroderma renal crisis was severe, progressive, and refractory to the standard of care therapy: oral captopril. Biopsy showed a diffuse thrombotic microangiopathy and findings consistent with scleroderma renal crisis. We hypothesize that depletion of systemic vascular endothelial growth factor with intravitreal anti–vascular endothelial growth factor injections likely contributed to the particularly severe presentation seen in this case. Though the finding of a monoclonal gammopathy of undetermined significance is another complicating factor, this case suggests that vascular endothelial growth factor inhibition may be a newly recognized trigger of scleroderma renal crisis.
Keywords
Introduction
Thrombotic microangiopathy (TMA) has been noted to develop in many disease states that have both inflammation and endothelial injury. 1 There are many complex pathways involved; however, it has become clear that endothelial injury can occur more easily in certain genetically susceptible individuals. 2 It has also been observed that if endothelial damage and complement activation are sufficiently severe, TMA can occur despite the appearance of an apparently benign genetic environment. 3 Scleroderma renal crisis (SRC) usually presents with a TMA that occurs within the context of malignant hypertension (MHT) and progressive renal dysfunction. 4
It is important to note that MHT is not always observed, and renal-limited TMAs related to SRC have been reported in individuals without findings of hypertension.5–7 The relationship between complement activation, rheumatologic syndromes, and endothelial dysfunction plays a key role in patients with MHT, SRC, and TMA. Timmermans et al. 8 recently reported the association between some genetic abnormalities to chronic hypertension with renal-limited TMA. Figure 1 explains the association between complement activation, genetic complement dysfunction, and endothelial shear stress that can create the conditions for a TMA to develop.

Schematic of causal theories of hypertension and thrombotic microangiopathy.
The use of high-dose corticosteroids is known to increase the risk of SRC, thus explaining why steroid therapy has fallen out of favor as a treatment of systemic sclerosis (Ssc). 9 The current treatment for SRC consists of renin angiotensin aldosterone system (RAAS) blockade and low dose aspirin for anti-platelet effect.10,11 The clinical benefit does not seem to be uniform, and there are no proven second- or third-line treatment options. 12 Experimental therapy with anti-endothelin agents, plasmapheresis, and complement blockade has been described for SRC. 13
Another agent recently noted to be systemically absorbed and proven to disrupt systemic vascular endothelial growth factor (VEGF) levels are intravitreal injections of VEGF inhibiting monoclonal antibodies.14–16 We present a case of SRC that initially developed after the patient was exposed to corticosteroids. He was subsequently exposed to intravitreal VEGF blockade and had rapidly progressive hypertension with subacute kidney injury refractory to the standard of care therapy. We thus bring attention to a possible role for VEGF blockade in accelerating SRC in this patient who received intravitreal injections of bevacizumab.
Case report
A 37-year-old male developed a sudden progressive onset of worsening skin thickening, initially localized to his torso but had since begun to spread. There was no cyanotic discoloration of extremities that would suggest Raynaud’s phenomenon. The patient also noted progressive weight loss up to 20–30 pounds (lbs) over the first 3 months of his initial presentation. The patient described fatigue, but without wheezing or dyspnea.
His systolic blood pressure was initially 100–110 mmHg, and he had no previous history of hypertension. His renal function was within normal range, with a serum creatinine of 0.8 mg/dL. Notably, he had a history of hepatitis B, treated with entecavir, without evidence of active disease (precluded use of B cell depletion therapy). His serological testing revealed a negative anti-nuclear antibody (titer of 0), negative anti-Smith antibodies, and negative anti-ribonucleoprotein. Anti-Scl-70 (anti-topoisomerase I antibody) levels were detected at a low level, but within the reference range (3–4 AU/mL). The triple negative profile was unusual, and as such more investigation was warranted before considering a diagnosis of Ssc.
A skin biopsy was sought from the patient’s left upper extremity at a site of severe skin thickening. The area of skin thickening was representative of changes over much of the patient’s skin surface, his modified Rodnan skin score was 31, and nail bed capillaroscopy exam was positive and suggestive of Ssc. There was no evidence of pericardial friction rub noted on exam, and echocardiography did not reveal pulmonary hypertension or a pericardial effusion.
The skin biopsy sample was read by the pathologist as suspicious for morphea versus scleroderma/Ssc. His renal function was normal until that time, and he had no gadolinium exposure. We clarified the point about gadolinium exposure to ensure that the patient did not have nephrogenic systemic fibrosis (NSF). The diagnosis of Ssc was more rigorously suspected on the basis of the skin biopsy results and careful clinical exam that revealed diffuse skin thickening and sclerodactyly. He was subsequently started on mycophenolate mofetil for treatment of Ssc.
A curious feature of his presentation was the finding of an IgG lambda monoclonal light chain of undetermined significance at 1.1 g/dL. A bone marrow biopsy was obtained and showed a normal cellular marrow, of which 10% were lambda restricted plasma cells, confirmed by flow cytometry. Cytogenetics/fluorescent in situ hybridization demonstrated a gain of 1q, gain of 5q, monosomy 13, and 14q minus ranging from 67% to 94%, karyotype was 46, XY.
Given the complexity of this case, he saw multiple providers at various health care centers. He was started on high-dose oral corticosteroids (60 mg), the reason for starting this agent was not immediately apparent to our team. Subsequently, he had a rise in blood pressure and serum creatinine from 0.8 to 1.1 mg/dL. This occurred abruptly after starting the prednisone, 3 months after his initial presentation. The patient’s blood pressure rose from 120–130 mmHg systolic baseline to 140–150 mmHg during this time.
He developed blurry vision and was referred to ophthalmology where dilated eye exam revealed “cotton wool spots.” He was given 1.25 mg intravitreally injected bevacizumab on two occasions. A week later, an even sharper rise in blood pressure was noted and accompanied by worsening serum Cr to 1.4 mg/dL. Figure 2 shows the trends of serum creatinine, systolic blood pressure, and diastolic blood pressures for this case. The data are presented in months after presentation to preserve anonymity.
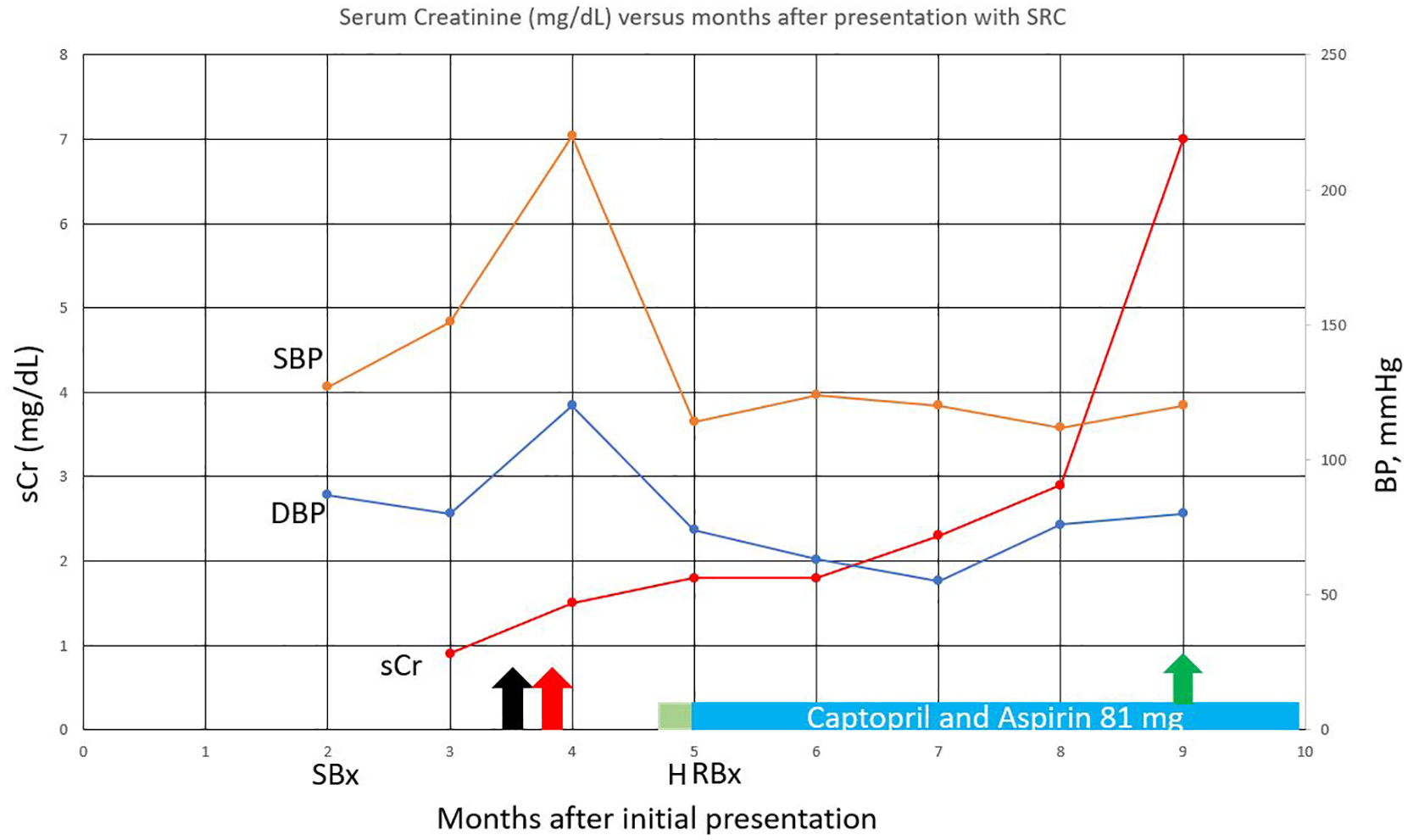
Serum creatinine (mg/dL) versus months after presentation with SRC.
He was admitted a week later with severe hypertension with systolic blood pressures of 200–220 mmHg. A marked increase in proteinuria from undetectable to a urine protein to creatinine ratio of 1-gram protein per gram creatinine was noted during this admission. A renal ultrasound and Doppler evaluation revealed normal sized kidneys without any evidence of atrophy or size mismatch, along with no evidence of renal artery stenosis. Given the sudden change in blood pressure, renal function, proteinuria, and recent exposure to oral corticosteroids, SRC was suspected. A complement-mediated TMA related to intravitreal VEGF inhibition could also not be ruled out. As such, a renal biopsy was obtained after his blood pressure was controlled to look for these diagnostic possibilities.
The patient’s renal biopsy (Figure 3) contained 57 glomeruli (1 globally sclerotic) and light microscopy demonstrated arteries with near-complete luminal occlusion by fibrointimal proliferation and adjacent ischemic tubulopathy. Mucoid intimal hyperplasia was noted in several arteries as well on trichrome stain, along with “onion skin” changes and bloodless glomeruli. Some rare glomeruli exhibit segmental small luminal thromboses. There was diffuse but early/evolving tubular atrophy as well as early interstitial fibrosis associated with the ischemic tubulopathy. Immunofluorescence was negative for any immune complex staining.

Features of active thrombotic microangiopathy on renal biopsy: (a) arteries with near-complete luminal occlusion by fibrointimal proliferation (arrows) and adjacent ischemic tubulopathy (periodic acid Schiff stain, 100×); (b) artery with mucoid intimal hyperplasia (arrow) (trichrome stain 200×); (c) artery with “onion skin” change (trichrome stain 400×); and (d) “bloodless” glomerulus (H&E stain, 400×).
A Congo-red stain was negative for amyloid deposition. Electron microscopic studies demonstrated marked ischemic-type corrugation of glomerular capillary loops as well as extensive endothelial cell swelling often with near-complete luminal occlusion. Glomerular subendothelial spaces were widened and showed occasional neomembrane formation. Luminal platelet aggregates were present. No electron dense, organized, or crystalline deposits were present in any compartment. The overall diagnosis was a diffuse TMA with arterial predominance, consistent with Ssc-associated renal disease.
The patient’s lactate dehydrogenase showed a low-level elevation at 344 IU/L, which is near the upper limit of normal. A disintegrin and metalloproteinase level motif #1, member 13 (ADAMTS13) level was 75%, ruling out ADAMTS13 deficiency and thrombotic thrombocytopenic purpura (TTP). International normalized ratio was normal at 1 with a normal prothrombin time of 10.5 s, and a normal activated partial thromboplastin time at 28 s. Platelets were normal ranging 300,000–358,000/µL, hemoglobin was 11.8 g/dL, and there was no evidence of microangiopathic hemolysis. Schistocytes were not noted during the patient’s course at any time. Reticulocytes were normal at 1.3%, iron saturation was 43%, and vitamin B12 level was 451 pg/mL. Shigatoxin was negative in the patient’s stool specimen. The data support a renal-limited TMA without peripheral hemolysis.
Serum VEGF level obtained weeks after injection with bevacizumab was 52 pg/mL. This level is near to lower limit of normal, and could suggest some level of VEGF depletion. The lack of an elevated level also rules out POEMS (polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes) syndrome which was in differential diagnosis, and is characterized by a high serum VEGF level.
Complement genetics were obtained and demonstrated a heterozygous, missense variant (c.1058C>T) of membrane cofactor protein (MCP) (alanine 353 to valine 353 substitution). This was previously associated with atypical hemolytic uremic syndrome (aHUS) presented in the case series of Fang et al. 17 in 2008. The risks and benefits of complement blockade were explained to patient, who was started on low dose 81-mg aspirin and captopril titration as inpatient and outpatient as the standard of care. The patient was made aware of the favorable response of eculizumab in 66.7% of SRC cases.18–24
The patient ultimately declined use of complement blockade despite the copious explanation of benefits, given the risks reported for meningitis despite existence of vaccines and prophylactic antibiotics. The patient’s renal function continued to worsen with serum creatinine rising to 1.9 mg/dL, to 2.7 mg/dL, and then to 7 mg/dL over the course of 5 months. Currently, he is receiving plasmapheresis to improve renal function, and renal replacement therapy. The plan is to pursue renal transplantation with complement blockade with a C5 or C3 blocking agent, depending on availability, and the patient’s willingness to try this adjunct therapy.
Discussion
Ssc-induced renal disease covers a wide spectrum, from a chronic interstitial nephritis with resulting mild proteinuria and indolent renal dysfunction, to the more severe complication of SRC. 12 The presentation of renal crisis includes severe hypertension with findings of end organ dysfunction with the risk of irreversible renal failure. 4 The pathological findings of SRC are generally an arterial/arteriolar predominant TMA often with marked subendothelial edema and fibrointimal proliferation resulting in prominent onion skinning–type change with or without concurrent glomerular thrombosis. 4
This particular patient’s presentation is unique in that a week before the patient was hospitalized with MHT, he received intravitreal VEGF injections for cotton wool spots in the eye by ophthalmology. There are now increasing reports of intravitreal injections resulting in VEGF depletion and glomerular disease including TMA, collapsing focal and segmental sclerosis (FSGS), as well as other causes of nephrotic syndrome.14–16,25–27 The renal biopsy findings we presented could be compatible with TMA from VEGF depletion and from SRC. Given the presence of Ssc clinically predating the intravitreal VEGF inhibition injections, it was surmised that the patient’s Ssc was likely exacerbated by VEGF blockade and hypertension as well as the oral corticosteroids.
Given the refractory nature of the TMA, the evidence for eculizumab was reviewed with the patient, but he felt that the risk of meningitis outweighed the potential benefit. In all, nine patients with Ssc syndromes were treated with eculizumab, with six patients showing a positive response. Table 1 details the cases of SRC treated with eculizumab and the corresponding clinical response data.18–24
Cases of eculizumab therapy in patients with TMA due to SRC.

CKD: chronic kidney disease; ESRD: end stage renal disease; F: female; M: male.
In addition, this patient’s genetic testing led to some equivocal findings of alleles not generally thought to be pathogenic. We cannot exclude a yet undescribed role for these genetic variants which affect susceptibility to complement amplifying and endothelium disruptive insults. It is probable that genetic susceptibility and trigger strength should both be taken into account when TMA episodes are observed. A “sliding scale” of genetic risk and trigger strength are likely operating concurrently, 28 with more purely genetic presentations resulting from minor environmental triggering exposures and more acquired presentations occurring from severe triggers. 1 See Figure 4 for a diagram illustrating this concept in TMA susceptibility.

The interplay between genetic risk and trigger strength in triggering thrombotic microangiopathy.
The patient’s monoclonal gammopathy also adds complexity to this case. TMA is considered to fall within the monoclonal gammopathy of renal significance (MGRS) spectrum of lesions. 29 Furthermore, paraneoplastic scleroderma like-tissue reactions have been reported. 30 The degree to which the patient’s underlying plasma cell dyscrasia may have influenced this patient’s clinical course are uncertain. The renal biopsy did not show any evidence for any other MGRS-associated lesion such as amyloidosis, light chain proximal tubulopathy, light chain cast nephropathy, light chain deposition disease, FSGS, or heavy chain deposition disease.31,32
Conclusion
We report a 37-year old male w/severe Ssc and the onset of SRC after oral corticosteroid treatment, and intravitreal VEGF blockade. More work remains to be done regarding the relationship of VEGF signaling and SRC. The effects of alternative complement pathway and VEGF blockade remain interesting due to the association of SRC with TMA. Oral corticosteroids should be avoided in Ssc, and we believe caution should be used when prescribing intravitreal VEGF blockade in patients with Ssc. Treatment with complement blockade needs to be thoroughly studied due to pathophysiological overlap between SRC and complement-mediated TMA.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Ramy Hanna discloses that he is a consultant for Alexion pharmaceuticals from 2018–2020, though this topic does not involve the use of complement blockade, it is discussed. Hence need for disclosure.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Ethical permission/consent for publication
IRB permission was not applied for as it is not required for individual case reports.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: I.B.K. is supported in part by funds from the NIH (R01-DK077162), the Allan Smidt Charitable Fund, the Factor Family Foundation, and the Ralph Block Family Foundation. K.K. is supported by the National Institutes of Health-National Institute of Diabetes, Digestive and Kidney Disease (NIH-NIDDK) grant K24-DK091419 as well as philanthropist grants from Mr Harold Simmons, Mr Louis Chang, Dr Joseph Lee, and AVEO.
Informed consent
We have retroactively obtained written informed consent that is required to publish patient information from patient, no images to be published.