
Abstract
Background:
Cancer drug development is a complex and costly process. Selinexor is a drug that received accelerated approval as a new treatment for relapsed or refractory diffuse-large B-cell lymphoma and multiple myeloma. Despite initially showing promise in treating these conditions, it has shown high-grade toxicity in clinical trials. Hence, an analysis is needed to assess the clinical trial portfolio of selinexor.
Objectives:
This investigation aims to evaluate published clinical trials of selinexor to assess its risk/benefit in terms of response and survival outcomes as well as its toxicity.
Design:
Cross-sectional.
Methods:
We conducted a cross-sectional investigation by searching databases for published clinical trials that used a response criteria pertaining to selinexor administration in adults. In a masked, duplicate manner, we extracted trial characteristics, median progression-free survival (PFS), overall survival (OS), objective response rates (ORR), and Grade 3–5 adverse events (AEs).
Results:
Of the 753 articles identified, 40 were included in our final sample. The trials reporting PFS data using control arms showed a median difference in PFS by 4.4 months, favoring the selinexor treatment arm. However, trials that reported OS data with control arms indicated that selinexor showed a worse median difference in OS (−2.4 months) than the control arms. Among the 53 measurements reporting ORR, the weighted median ORR was 36.4%, and the median difference ORR (4.8%) favored selinexor. Additionally, 4153 cumulative Grade 3–5 AEs were reported.
Conclusion:
In comparison to a control arm, selinexor increases PFS and induces response, suggesting drug activity. However, acceptable Grade 3–5 AEs or improvement of OS was not seen across a single indication, suggesting a poor pretest probability. Our risk/benefit analysis of selinexor provides valuable insight into the unfavorable outcomes of the drug and increased high-grade AEs. Hence, further testing of selinexor should be carefully scrutinized and contextualized with the portfolio of data we present.
Introduction
The development of novel cancer drugs is a complex and precarious process with the potential to deliver massive benefits to both patients and drug manufacturers.1–3 Cancer drugs are expensive to develop, often costing billions of dollars in research and development before regulatory approval. 4 These drugs also take significant time to develop—an average of about 7.2 years from initial testing to Food and Drug Administration (FDA) approval. 5 Drug manufacturers take significant risks with the development of these novel cancer drugs, often spending millions of dollars upfront for research and development. 6 Should one of these novel cancer drugs fail to achieve regulatory approval, the manufacturers risk significant financial losses. On the contrary, after successful initial regulatory approval, further testing for additional indications (e.g., other types of cancer) may be a source of additional revenue and market share for drug manufacturers.
When drug manufacturers explore additional indications for their drug, they may threaten the causal inference of their clinical trials. Multiplicity, or multiple outcomes testing, increases the risk of false positive results and may lead to an erroneous outcome in a clinical trial. 7 The phenomena of multiplicity has been studied in several cancer drugs including bevacizumab, 8 sunitinib, 9 and imatinib. 10 The potential harm of multiplicity in cancer drug development is best illustrated by sunitinib. Carlisle et al. 9 showed that after initial regulatory approval in 2006 for renal cell carcinoma and gastrointestinal stromal tumors, sunitinib was tested over 100 times for additional indications with a cumulative analysis demonstrating persistently increasing Grade 3–5 adverse events (AEs; which includes treatment-related deaths) and decreasing response rates. They concluded that the portfolio of sunitinib after initial approval demonstrated increased harm and decreased benefit to patients.
Recently, selinexor received accelerated approval for the treatment of relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and multiple myeloma. Selinexor is a first-in-class inhibitor of the exportin-1 oncoprotein, which is correlated with poor patient outcomes and treatment resistance.11,12 The excitement surrounding selinexor for both indications centered on the idea that selinexor was providing an additional treatment option to patients with heavily pre-treated disease.13,14 For multiple myeloma, specifically, it is standard of care to give patients upfront triplet therapy, including a proteasome inhibitor (e.g., bortezomib), immunomodulatory agent (e.g., lenalidomide), and dexamethasone, or quadruplet therapy, 15 which adds the CD38 inhibitor daratumumab. If a patient’s myeloma progresses and becomes refractory to these frontline drugs, outcomes are notably poorer and treatment options are more limited.16,17 Therefore, a novel agent in this heavily pre-treated setting offers the hope of longer survival and improved quality of life late in the disease process. However, in both of the trials leading to FDA approval for selinexor, there was a signal for significant high-grade toxicity that may affect the risk/benefit calculus for these drugs in patients who already have a high burden of resistant disease.
The goal of this investigation is to conduct a cross-sectional analysis of the published clinical trials for selinexor to characterize its benefit to patients (in terms of response and survival outcomes) and toxicity (in terms of drug-related AEs) across all indications. The rationale for this analysis was to rationalize and contextualize future testing of selinexor should the cumulative risk/benefit ratio have suggested a poor pretest probability of success for other indications.
Methods
Ethical oversight
The Oklahoma State University Center for Health Sciences reviewed our protocol and determined that this research qualifies as non-human subjects research as defined in regulation 45 CFR 46.102(d) and (f).
Study design/open-science
This was a cross-sectional study of selinexor (Xpovio®, Karyopharm Therapeutics) clinical trials and its risk/benefit profile throughout development and applications to indications outside of initial approval. To improve reproducibility and further enhance academic rigor, a protocol was uploaded prior to the investigation. Raw data, statistical analysis scripts, protocol, and extraction forms have been made publicly available via the Open Science Framework (OSF). 18
Research questions, definitions, and hypothesis
We aimed to assess the following questions throughout our study: (1) given the high cost and hazards patients may experience in clinical trials, what are the risk/benefit profiles of clinical trials assessing the efficacy and safety of selinexor? (2) Does the combined risk/benefit portfolio of selinexor favor increase overall risk to patients? In our study, we defined a clinical trial profile as the overall risk and benefit experienced by participants within a single trial as measured by methods and standards mentioned in our “Data extraction” section. The portfolio of a drug was defined as the total trial profiles for a given intervention. We hypothesized that the analysis of the cumulative risk–beneft ratio of selinexor would demonstrate an increased number of negative outcomes associated with higher patient risk, illustrating an overall negative drug portfolio.
Training
The authors were trained on clinical trial design, responses, and outcomes by clinical oncologist Vinay Prasad (V.P.), an expert in evidence-based medicine. Authors were also educated on both the Common Terminology Criteria for Adverse Events (CTCAE) 19 and the International Myeloma Working Group Uniform Response Criteria (IMWG-URC). 20 Additionally, authors were also trained on other response criteria that arose within the sample. A full list of these specific response criteria used in trials other than RECIST can be seen in the Supplemental Material. Screening authors were trained to use the online systematic review platform Rayyan (https://www.rayyan.ai/). 21 Data extraction was facilitated through the use of a pilot-tested Google Form. Before extracting our sample for the study, authors were trained using five sample studies, which will ensure consistency.
Literature search
On May 25, 2023, we searched PubMed/MEDLINE, Embase (Elsevier), Cochrane (CENTRAL), and ClinicalTrials.gov for clinical trials that tested selinexor as monotherapy or in combination with other pharmaceutical interventions for cancer treatment. We used the PolyGlot Search Translator, 22 developed by Bond University and the Institute for Evidence Based Healthcare, to standardize our use of search strings across multiple databases. 21 All search strings—including the initial returns and date of search—were uploaded to OSF. 18
Selection process
We uploaded search returns into Rayyan for literature screening. In a masked duplicate fashion, three investigators (Annes Elfar (A.E.), Andrew V. Tran (A.V.T.), and Joseph Case (J.C.) screened titles and abstracts for potential inclusion. After completing the screening, a fourth investigator Ryan McIntire (R.M.) was available to resolve any discrepancies. All exclusion reasons during the screening process were documented and used to create a PRISMA flowchart of exclusion decisions.
Inclusion and exclusion criteria
The following inclusion criteria were applied: (1) human adult clinical trial studies, (2) evaluation of the efficacy of selinexor as monotherapy or in combination with other treatment regimens, (3) assessment of the benefit of selinexor using radiographically derived response criteria for solid tumors (e.g., RECIST) and laboratory-derived response criteria for non-solid tumors, and (4) a publication in English. We excluded study designs that did not meet our inclusion criteria such as non-oncological studies, biosimilars, studies on healthy individuals, and non-manuscript publication types including: secondary reports, interim results, clinical trial updates and follow-ups, preclinical studies, literature reviews, systematic reviews, meta-analyses, human tissue studies, laboratory studies, case reports, letters, editorials, opinion pieces, conference abstracts, corrections, and redactions.
Data extraction
After data screening, a final study pool underwent data extraction in a masked, duplicate fashion by two authors (A.E., A.V.T.) with a third author (R.M.) available to resolve discrepancies. Authors extracted the following trial characteristic variables, which include: published trial title, PubMed ID, clinical trial registry number, the affiliated country of the first author, date of publication, number of participants, mean or median age of participants, number of male participants, number of female participants, trial indication(s), metastatic or non-metastatic stage, the median or mean age of trial participants in one or more arms, if the trial was controlled, whether the trial assessed monotherapy or combination therapies, study sponsor including funding, number of centers, blinding of the trial participants, randomization ratio, type of analysis, phase of trial, conflicts of interest statements, and if the trial was negative, indeterminate, or positive. A trial was deemed negative if it did not meet its pre-specified endpoints or was excessively toxic, indeterminate if it did not pre-specify endpoints and was using a tolerable regimen, or positive if it met its pre-specified endpoints using a tolerable regimen. The tolerability of a regimen was determined by trial authors. 9
The following variables were extracted for risk and benefit outcomes from treatment arms: the name of the arm, the number of participants for grade assessment, AEs grade, median progression-free survival (PFS) in months, median overall survival (OS) in months, partial response rate, complete response (CR) rate, objective response rate (ORR) (defined by the RECIST or similar criteria), and number of Grade 3–5 treatment-related AEs. Outcome measurements and AEs of pre-specified indications compassing all trial participants were extracted.
We made several decisions about trial characteristics to ensure consistent and reliable data extraction. Trials with one or more phases were recorded as the higher phase. If confirmed or unconfirmed responses were specified by trialists, we only extracted the confirmed responses; if measurement confirmation was conducted by independent investigators as specified by trialists, we extracted the independently confirmed measurements. We pooled dose-escalation, dose-expansion treatment, and indication arms into individual summary arms. We extracted variables of interest from the pre-crossover allocation groups to control for carryover effects that might interfere with the response rate in crossover trials. If a trial enrolled participants in more than two indications, we reported those as umbrella trials. Basket trials of indications of similar mutations were reported by their mutation classification. Lastly, all participants were included in calculations of ORRs of each arm unless trialists specified evaluable patients.
Statistical analysis
We conducted descriptive statistics in R and RStudio (Posit Workbench, Posit, PBC) (version 4.2.1) to analyze the weighted median proportion of ORR by trials and indications as well as CR by trials. Additionally, Lucidchart software was used to construct an AERO diagram, representing phases of trials across different indications over time, which can be seen in Figure 1.

AERO diagram demonstrating included trials published over time (n = 40); the legend shows phase of the trial and whether it was deemed positive, indeterminate, or negative.
Results
Our initial database search identified 753 articles—of which, 40 were selected and included in our final analysis (Figure 2). In 70% of the trials (23/40), selinexor was administered in combination with another drug. Nearly all of the trials of the selinexor (39/40; 98%) received at least partial funding or support from the pharmaceutical industry.

PRISMA diagram depicting included and excluded studies.
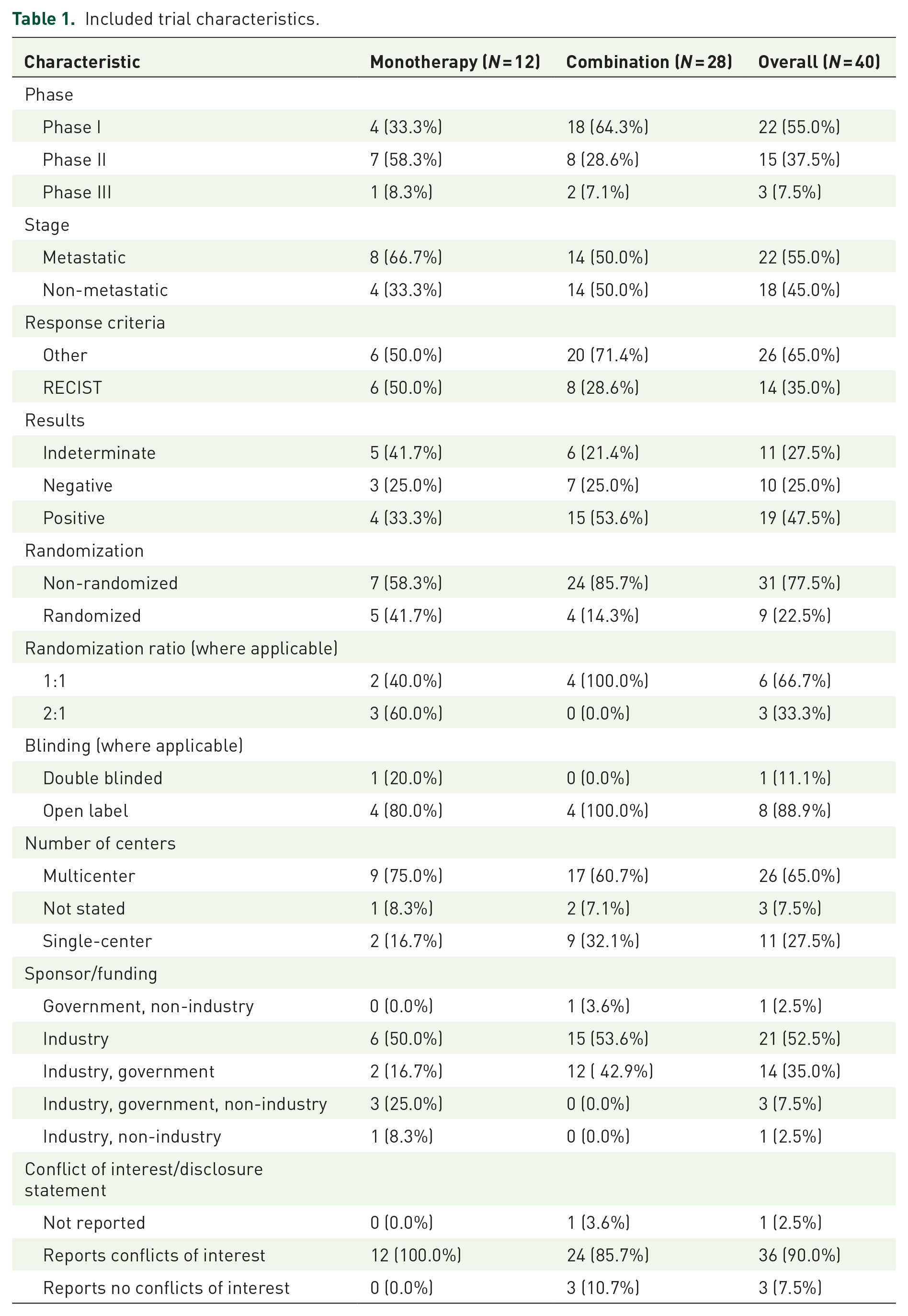
Overall, 65% of the trials (26/40) in our sample were multicenter trials, and the majority were non-randomized clinical trials (31/40; 77.5%). Over half of the trials included in our sample were phase I trials (22/40; 55%), followed by phase II (15/40; 37.5%), and the least being phase III (3/40; 7.5%). A majority of trials (36/40; 90%) included conflicts of interest statements in their primary publication. Fourteen trials (14/40; 35%) used the RECIST response criteria, while 26 trials (26/40; 65%) used various response criteria relevant to their respective indications. A total of 19 trials (19/40; 47.5%) were deemed positive by trialists, 10 trials (10/40; 25%) were considered negative, and 11 trials (11/40; 27.5%) were considered indeterminate. Additional information regarding trial characteristics is highlighted in Table 1.
Included trial characteristics.
The median PFS of selinexor regimens across all trials (n = 40), comprising 34 measurements, was 2.9 (IQR, 2.3–8.15) months. The median OS of selinexor regimens, comprising 36 measurements, was 9.2 (IQR, 6.5–12.0) months. There were four comparative PFS measurements across three trials (3/40; 7.5%), and three comparative OS measurements across three trials (3/40; 7.5%). In the three trials reporting PFS, the median delta PFS was 4.4 (IQR, 0.7–7.2) months in favor of selinexor. Conversely, for OS the median delta OS was −2.4 (range −2.9 to −2.0) months in favor of the control arms. Selinexor demonstrated a worse median OS than the control arm in all three trials (Table 2).
ΔPFS and ΔOS for randomized controlled trials.
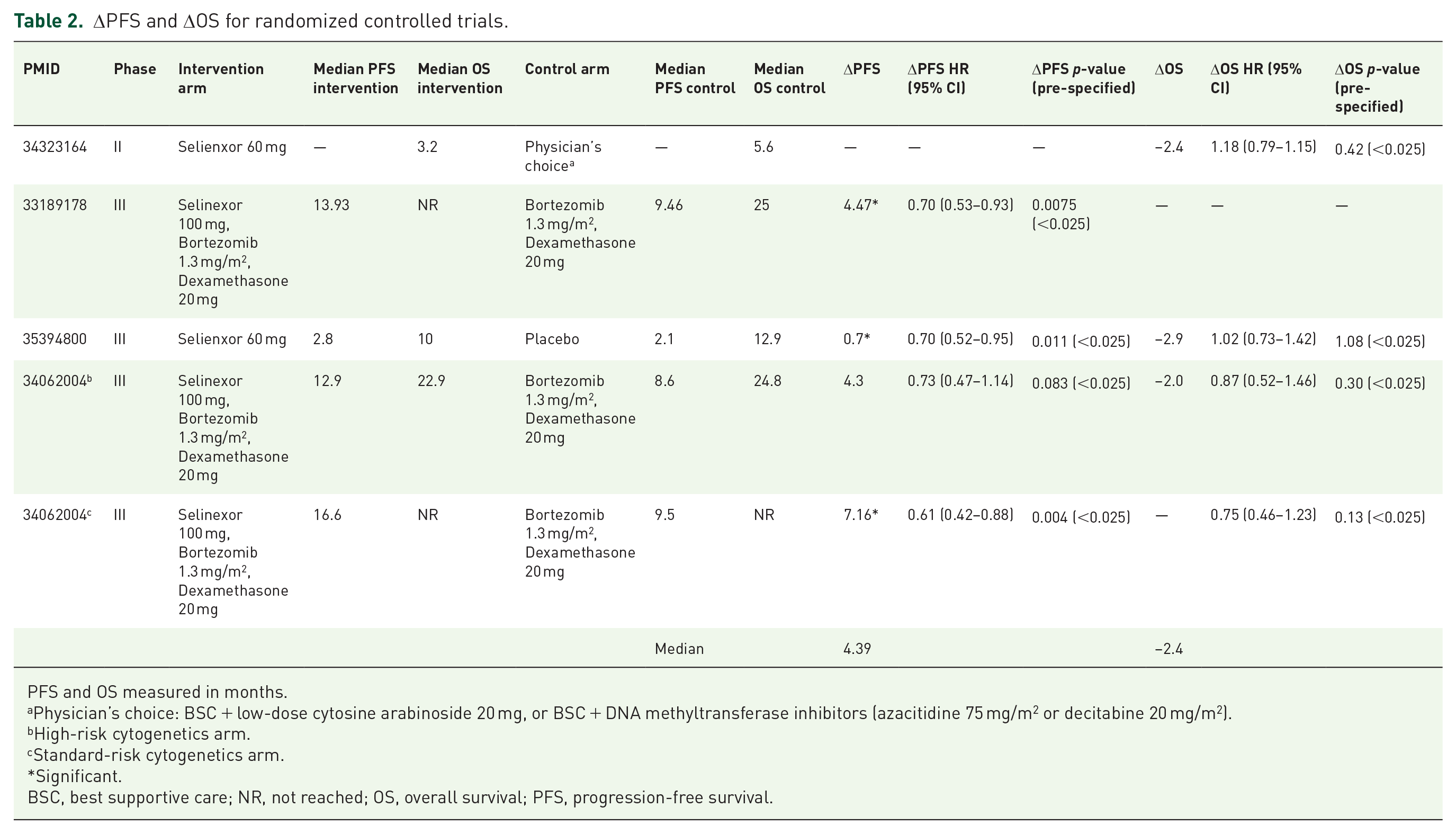
PFS and OS measured in months.
Physician’s choice: BSC + low-dose cytosine arabinoside 20 mg, or BSC + DNA methyltransferase inhibitors (azacitidine 75 mg/m2 or decitabine 20 mg/m2).
High-risk cytogenetics arm.
Standard-risk cytogenetics arm.
Significant.
BSC, best supportive care; NR, not reached; OS, overall survival; PFS, progression-free survival.
There were 53 reported measurements of ORR for selinexor regimens with a weighted median ORR of 36.4% (95% CI, 28.8%–43.9%; Figure 3). For the 50 measurements of CR, the weighted median was 14.3% (95% CI, 9.1%–19.6%; Figure 4). Three trials reported ORR for a control arm and the median delta ORR was 4.8% (range, 2.7%–14.1%) in favor of selinexor. Four trials reported CR of a control arm and the median delta CR was 3.1% (range, −5.4% to 6.8%). An analysis of weighted ORR by specific indication is shown in Figure 5.

Forest plot depicting objective response rate by trial (n = 40).
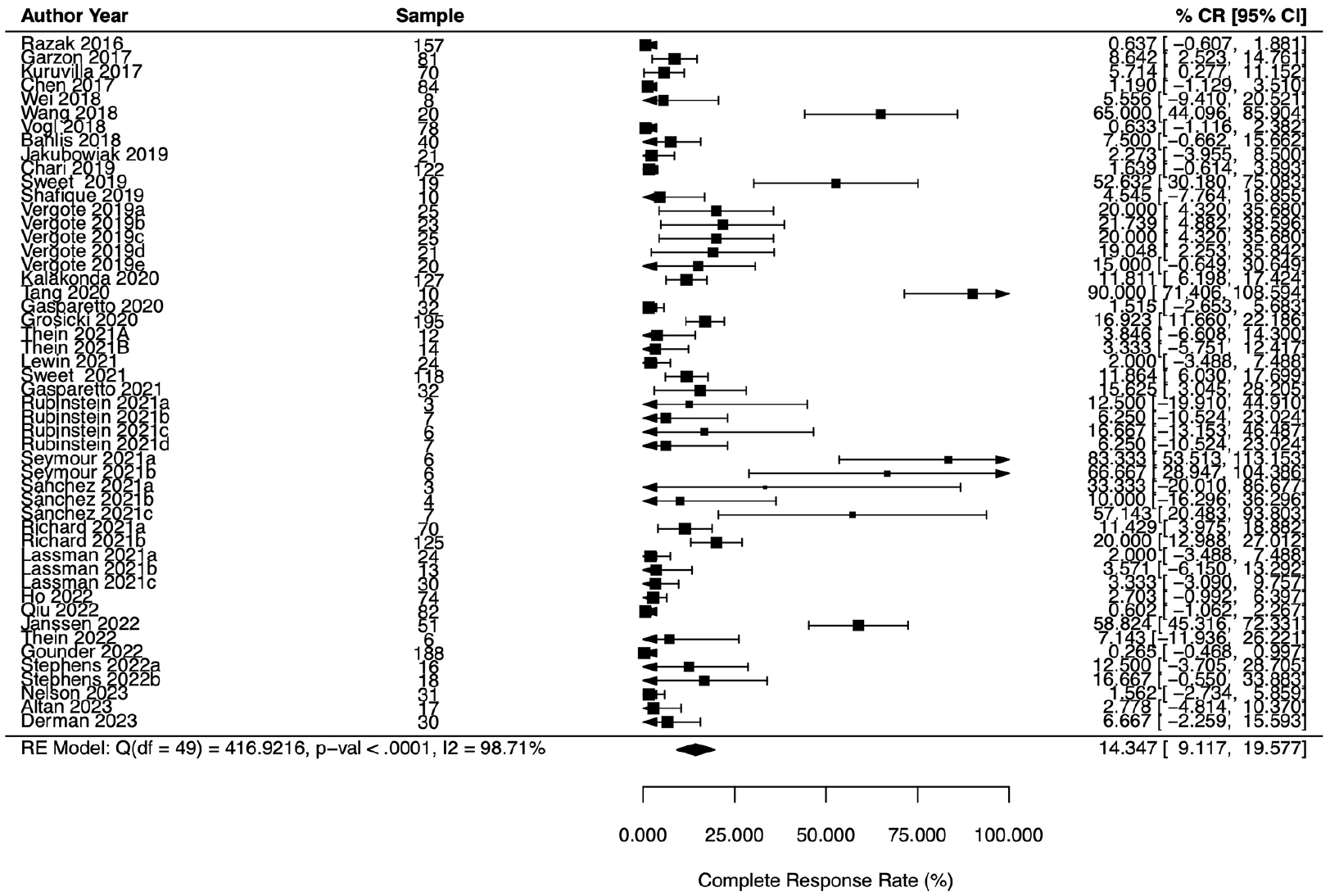
Forest plot depicting complete response by trial (n = 40).

Forest plot depicting objective response rate by indication.
With respect to Grade 3–5 AEs, there were 4069 Grade 3–4 AEs and 84 Grade 5 AEs reported, amounting to a total of 4153 cumulative AEs across all trials and a median 1.5 (IQR, 1.0–1.8) Grade 3–5 AEs per patient enrolled. The indication with the most enrolled patients was multiple myeloma with 10 trials and 832 patients—these patients experienced 2.1 AEs per patient. Further trial characteristics and AE by indication can be seen in Table 3. Cumulative Grade 3–4 and Grade 5 AE rates can be seen in Figure 6. Among the trials that compared selinexor with a comparative control arm (5/40; 12%), there were 1049 Grade 3–4 and 64 Grade 5 AEs reported in selinexor arms, as opposed to the 503 Grade 3–4 and 30 Grade 5 AEs reported in the control arms. Further details regarding AEs reported by trials with comparative control arms may be seen in Table 4. Of the 40 trials in our sample, 462 patients withdrew or were discontinued from selinexor after experiencing Grade 3–4 AEs, a median of 5 (IQR, 2.0–14.0) patients.
Characteristics and AEs by indication (n = 40 trials).
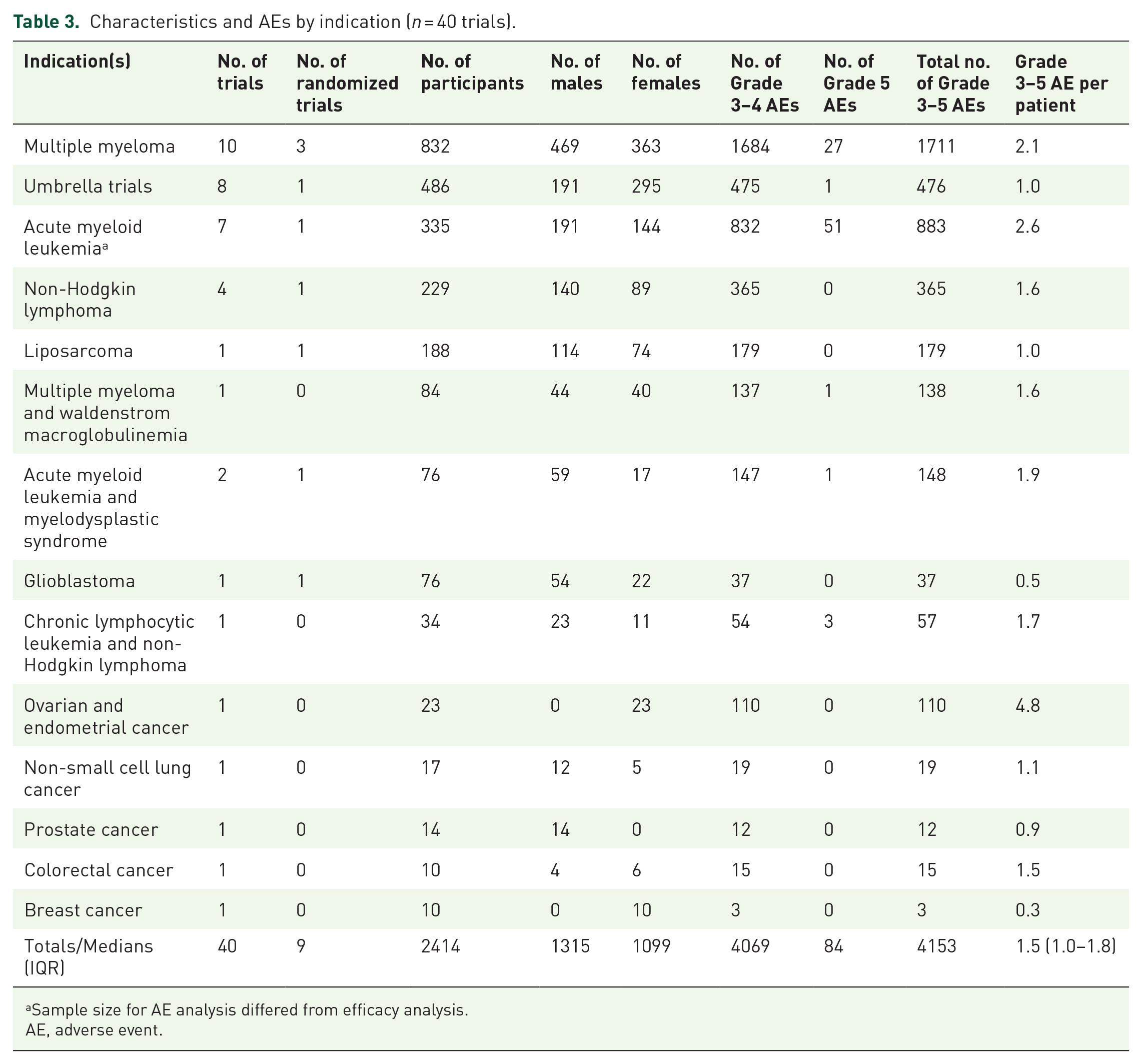
Sample size for AE analysis differed from efficacy analysis.
AE, adverse event.

Cumulative Grade 3–4 and cumulative Grade 5 adverse event rates.
AEs reported by trials with comparative control arms.

Arm 1 consisted of treatment with selinexor while Arm 2 consisted of the comparative control arm within the corresponding trial.
Dose reduction, initial dosage selinexor 55 mg/m2.
Physician’s choice: BSC + low-dose cytosine arabinoside 20 mg, or BSC + DNA methyltransferase inhibitors.
AE, adverse event; BSC, best supportive care.
Selinexor dosing regimens across studies exhibited heterogeneity, with initial doses, dose escalations, dose expansions, dose reductions, and dose modifications ranging from 20 mg to 100 mg and schedules varying from daily to biweekly; 60 mg taken twice a week was the most common regimen. Due to the variability in dosing schedules and their frequent dose reductions in response to AEs, we are unable to aggregate AEs across doses and schedules. Additionally 25/40 (62.5%) trials did not fully report all Grade 1–5 AEs.
Discussion
This investigation of the benefits and risks of selinexor across its portfolio of clinical trials found that selinexor has evidence of drug activity, but has to date failed to improve OS and confers significant high-grade toxicity to patients. Specifically, we found that compared to control arms, selinexor extends PFS and is able to induce response in about 36% of patients, about one-third of which achieve remission. However, the OS and AE data are not as favorable. Patients who received selinexor lived a median of 2.4 months less than patients in control arms, and across all trials of selinexor we found that there was a median of 1.5 high-grade AEs per patient. Taken together, our findings raise questions about whether further testing of selinexor in future trials is justified.
Our study raises several questions about selinexor with regard to its overall benefit to patients. Our results show that selinexor is able to demonstrate anti-cancer activity in various types of solid and hematologic cancers; however, we were never able to identify an indication with adequate sample size where AE rates were acceptable or OS was improved. One may argue that inducing a response is valuable to a patient whose cancer is already refractory to several frontline therapies. A partial or complete remission may allow a patient more time at home, rather than in the hospital or infusion clinic. 23 However, as our data show, this hypothetical time at home offered by the anticancer activity of selinexor may be counterbalanced by Grade 3 or 4 toxicities that often require hospitalization. Altogether, our data suggest that the pretest probability for selinexor improving OS is extremely poor, whereas the likelihood of conferring numerous Grade 3–5 toxicities is high. Ongoing monitoring of toxicity profiles across trials could guide adaptive dose regimens.
The heterogeneity observed in the dosing regimens of selinexor reflects the challenges of optimizing treatment strategies for a drug with a narrow therapeutic index, particularly when Grade 3–5 AEs remain noteworthy contributors to treatment discontinuation. In clinical trials, the term “tolerability” is often determined by trialist criteria, which may standardize AE reporting but sometimes present a limited view of patient experiences. For selinexor, tolerability is particularly relevant given its weekly dosing schedule without planned breaks unless AEs necessitate dose modifications. This contrasts with traditional cytotoxic chemotherapy regimens, which are often administered every 2 or 3 weeks, allowing for more extended periods without side effects. Consequently, even recurrent Grade 2 AEs, such as fatigue or nausea, may cumulatively impose a significant burden on patients receiving selinexor, potentially making the treatment intolerable from their perspective. Therefore, tolerability as defined by trial criteria may not fully capture AEs from a patient-centered perspective. Future trials and post-marketing studies could benefit from incorporating patient-reported outcomes and quality of life assessments to provide a more comprehensive view of tolerability.
Selinexor does appear to have activity against myeloma. However, much of this is from early-phase trials and our dataset only reports three phase III trials of selinexor. Previous studies have shown an overestimation of early-phase efficacy in the sense that early-phase trials frequently report higher efficacy rates than more advanced phases. For example, in PD-1/PD-L1 inhibitor trials, early phases reported significant response rates that were not consistently replicated in phase III trials, likely due to smaller sample sizes, selective patient criteria, and potential early-phase biases. 24 This trend, sometimes referred to as “early-phase optimism,” may reflect exaggerated initial outcomes that skew clinical expectations. Given the high proportion of early-stage trials of the sample, these biases could potentially influence the perceived efficacy of selinexor.
The results demonstrated by selinexor are not too different from several other recent, controversial FDA drug approvals. In recent years, there have been several “dangling” FDA approvals. Dangling approvals occur when a drug receives accelerated approval based on a surrogate endpoint (e.g., ORR or PFS), but the postmarket trial fails to demonstrate favorable, patient-centered results (e.g., OS). In other words, these drugs with dangling approvals were able to demonstrate drug activity, but not drug efficacy. Ten such dangling approvals were identified in a recent analysis published in 2021. 25 The drug companies voluntarily withdrew four of these drugs, but of the six that went to the Oncology Drug Advisory Committee, only two were withdrawn. The implication is that despite 10 drugs failing to meet a priori established postmarket criteria, only 6 drugs are now considered unsafe for patients. This is the setting in which selinexor obtained accelerated approval in multiple myeloma and DLBCL. The postmarket trials have not yet been published for either criteria; however, our data suggest that in comparison to control arms, selinexor is unlikely to significantly improve OS.
Prior investigations similar to ours have assessed the portfolio of other cancer drugs.8–10 These analyses, as well as ours, indicate a common theme in oncology drug development. It would seem that drug manufacturers know, or have a good pretest probability, for which indications are best to test their novel drug. With respect to selinexor, multiple myeloma was one of the first indications in which selinexor was tested. However, after initial success, the drug manufacturers begin to probe other indications, testing to see if their drug will be effective and garner additional market share. We believe this strategy is reasonable for any company and should not be discouraged. We would, however, ask at what point does the probing stop? At what point does the pretest probability of success tip in a direction that is not favorable to patients? In the case of selinexor, we are unable to find one instance where it is able to improve survival for patients, yet we consistently see that it has high-grade toxicities. Moreover, the principle of multiple outcomes testing suggests that the likelihood of a false positive outcome increases the more testing occurs. Based on our interpretation of the data, we believe that further testing of selinexor should be approached with caution and carefully considered in the context of the data we present.
Our study had many notable strengths with some limitations. First, we implemented a broad clinical question and included all human trials of selinexor by searching multiple trial registries. 26 In doing so we were able to capture data from phase I to phase III for a range of outcomes from response rate to OS. Secondly, to minimize bias and increase transparency, our study was conducted using a masked-duplicate fashion in accordance with current Cochrane guidelines. 27 Additionally, in an attempt to increase the reproducibility of our study, our research methods and raw data were made publicly available. Furthermore, we also used an accepted methodology from previous research to augment validity.9,10 Limitations of our study include the lack of generalizability of our sample across all clinical trials of oncological interventions. Moreover, our sample size was relatively small, considering that many studies did not fit our inclusion criteria. Given the significant heterogeneity in dosing regimens and patient discontinuations due to toxicity within our sample, we were unable to meaningfully aggregate AE data across doses and schedules. It is possible that our systematic search may have not returned every trial pertinent to our study, known to be a common limitation of systematic reviews. 28 Despite various measures taken to maximize accuracy, potential errors within our extraction data extraction are still possible. Future studies could expand on our findings by incorporating reported toxicity data from publicly available patient registries, which may provide a more comprehensive understanding of the safety profile of selinexor in clinical practice beyond the controlled settings of clinical trials.
Overall, our data suggest that the pretest probability of selinexor improving OS or quality of life in a new indication is poor. This low pretest probability is based on our analysis of 40 selinexor trials which showed consistent evidence of drug activity, but no evidence of improved OS and significant, high-grade toxicity. Hence, thorough evaluation and contextual consideration of our findings are necessary prior to future trials of selinexor.
Supplemental Material
sj-docx-1-tah-10.1177_20406207251329174 – Supplemental material for An evaluation of selinexor’s clinical trial portfolio: a cross-sectional study
Supplemental material, sj-docx-1-tah-10.1177_20406207251329174 for An evaluation of selinexor’s clinical trial portfolio: a cross-sectional study by Annes Elfar, Andrew V. Tran, Joseph Case, Cole Wayant, Griffin K. Hughes, Ryan McIntire, Brooke Gardner, Chase Ladd, Andriana M. Peña, Jordan Tuia, Alyson Haslam, Vinay Prasad and Matt Vassar in Therapeutic Advances in Hematology
Supplemental Material
sj-docx-2-tah-10.1177_20406207251329174 – Supplemental material for An evaluation of selinexor’s clinical trial portfolio: a cross-sectional study
Supplemental material, sj-docx-2-tah-10.1177_20406207251329174 for An evaluation of selinexor’s clinical trial portfolio: a cross-sectional study by Annes Elfar, Andrew V. Tran, Joseph Case, Cole Wayant, Griffin K. Hughes, Ryan McIntire, Brooke Gardner, Chase Ladd, Andriana M. Peña, Jordan Tuia, Alyson Haslam, Vinay Prasad and Matt Vassar in Therapeutic Advances in Hematology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.