
Abstract
People with severe hemophilia A usually experience their first bleed early in life. In children with severe hemophilia A, primary prophylaxis is recommended to prevent recurrent and potentially life-threatening bleeds that significantly impact day-to-day life. Factor VIII (FVIII) prophylaxis is well-established in children and has been shown to reduce the development of hemophilic arthropathy. However, a major challenge of FVIII therapy is the development of neutralizing anti-FVIII antibodies (FVIII inhibitors). Simoctocog alfa (Nuwiq®) is a human cell line-derived recombinant FVIII (rFVIII) whose immunogenicity, efficacy, and safety have been studied in 167 children with severe hemophilia A across two prospective clinical trials and their long-term extensions. In 105 previously untreated children, the inhibitor rate of 16.2% for high-titer inhibitors (26.7% for all inhibitors) was lower than published rates for hamster cell line-derived rFVIII products. There was no inhibitor development in previously untreated children with non-null F8 mutations and in previously treated children. In a case series of 10 inhibitor patients, 8 (80%) underwent successful immune tolerance induction with simoctocog alfa with a median time to undetectable inhibitor of 3.5 months. In an analysis of 96 children who enrolled in the extension studies and received long-term simoctocog alfa prophylaxis for up to 5 years, median spontaneous, joint, and total annualized bleeding rates were 0.3, 0.4, and 1.8, respectively. No thromboembolisms were reported in any of the 167 children, and there were no treatment-related deaths. Optimal care of children should consider several factors, including minimization of inhibitor development risk, maintaining tolerance to FVIII, highly effective bleed prevention and treatment, safety, and impact on long-term outcomes such as bone and joint health. In this context we review the pediatric clinical data and ongoing studies with simoctocog alfa.
Keywords
Introduction
Hemophilia A is commonly diagnosed during early childhood, and most individuals with severe hemophilia A experience bruising or mucosal bleeds as first bleeding symptoms.1,2 Diagnosis in newborns is rare unless a positive family history is known or a severe, often life-threatening, bleed occurs (i.e. intracranial hemorrhage, severe bleed after venipuncture, or invasive procedures).1–4 In addition to the challenges of the bleeds themselves, repeated bleeds into joints in early childhood can lead to the development of hemophilic arthropathy, a common and debilitating complication in people with hemophilia A. 5 Furthermore, asymptomatic, or ‘silent’, bleeds into the joints may contribute to the development of arthropathy despite the absence of clinically evident joint bleeds. 5 For all these reasons, early initiation of prophylaxis is of utmost importance in children with severe hemophilia A.
Replacement FVIII therapy has been used for many decades to correct the underlying FVIII deficiency in hemophilia A patients.6,7 FVIII therapy is administered as prophylaxis to prevent bleeding, on-demand to treat breakthrough bleeds, or as surgical prophylaxis to provide perioperative hemostatic cover. 6 The effectiveness and safety of FVIII therapy have been well documented, and FVIII prophylaxis represents the standard of care for children with severe hemophilia A.8,9 Effective prophylaxis offers children the opportunity to participate in regular daily activities and live a healthy life.6,9 The most significant limitation of FVIII therapy is the development of neutralizing anti-FVIII antibodies, usually referred to as FVIII inhibitors, that increase the health and economic burden of hemophilia A.10–14 FVIII inhibitors are estimated to develop in up to 40% of previously untreated patients (PUPs) with severe hemophilia A, and minimization of this risk is an important treatment consideration.15,16 Immune tolerance induction (ITI) is the only clinically proven strategy for eradication of inhibitors and is recommended as the primary treatment option for patients with inhibitors.6,15,17,18 ITI involves prolonged intensive treatment with FVIII to eliminate FVIII inhibitors and has a success rate of 60–80%.15,19,20
More recently, emicizumab (Hemlibra®), a bispecific recombinant monoclonal antibody that mimics the co-factorial function of activated FVIII, has been licensed for prophylaxis in hemophilia A patients with and without FVIII inhibitors.21,22 Several other non-factor therapies are in development and gene therapy is licensed in adults with hemophilia A.7,23,24. Emicizumab is not indicated for acute bleeding control or surgical prophylaxis, and in such settings, concomitant hemostatic therapy is required. 25 In non-inhibitor patients on emicizumab prophylaxis, the need to minimize the risk of developing FVIII inhibitors, maintain achieved tolerance, and provide effective FVIII treatment remains.25–27
Simoctocog alfa (Nuwiq®) is a B-domain-deleted recombinant FVIII (rFVIII), without protein fusion or chemical modifications, produced in a human cell line.28–30 Based on the data obtained from the comprehensive clinical trial program in both adults and children with severe hemophilia A, it is indicated for the treatment and prevention of bleeds in people with severe hemophilia A of all ages. 31 Here, we describe the key findings from the simoctocog alfa clinical trial program in children with severe hemophilia A and describe ongoing real-world and scientific studies with simoctocog alfa that may offer further insight into the management of children with severe hemophilia A.
Clinical trials with simoctocog alfa in children
Two phase III studies32,33 of simoctocog alfa, each with an optional extension study,34,35 were conducted in children with severe hemophilia A, one in previously treated patients (PTPs)32,34 and one in PUPs.33,35 Both studies were prospective, international, open-label, non-controlled studies that investigated the efficacy, immunogenicity, and safety of simoctocog alfa. GENA-03 enrolled 59 PTPs [who had at least 50 previous exposure days (EDs) to another FVIII concentrate] aged 2–12 years at 15 sites in 7 countries. 32 The NuProtect (GENA-05) study enrolled and treated 108 PUPs of any age at 38 sites in 17 countries. 33 Patients who completed these studies were offered the opportunity to enroll in the respective long-term extension studies GENA-13 34 and GENA-15 35 (NuProtect-Extension). Details of the study designs have been published previously.32–36 All children in GENA-03, GENA-13, and the NuProtect-Extension study received prophylaxis, whereas children (PUPs) in NuProtect received prophylaxis or on-demand treatment at the discretion of the treating physician. In total, 167 children aged 0.7–12 years at first treatment were treated with simoctocog alfa.
Immunogenicity of simoctocog alfa in PUPs
When starting treatment in PUPs, minimizing the risk of the development of FVIII inhibitors is an important concern for clinicians, patients, and their families. Inhibitors develop from a multicausal immune response involving both genetic (unmodifiable) and environmental (modifiable) factors, only in part identified.37,38 The type of F8 gene mutation is the most important known genetic risk factor for inhibitor development, with null mutations being associated with the highest risk. 37 Several modifiable factors have been suggested to influence the risk of inhibitor development, including intensity of treatment and choice of FVIII product.37,38
The NuProtect study investigated the immunogenicity of simoctocog alfa in true PUPs, that is, patients who had no previous exposure to an FVIII concentrate or other FVIII-containing blood product. 36 Immunogenicity was assessed by the incidence of inhibitor development. In patients with severe hemophilia A, most inhibitors develop within the first 20 EDs, with a residual risk up to 75 EDs. 39 In the NuProtect study, patients were treated for 100 EDs or up to 5 years, whichever came first, and an intensive testing schedule was employed to capture the incidence and timing of inhibitor development.
A total of 108 children were included in the NuProtect study (median age of 12 months at first treatment), making it the largest study to prospectively investigate the immunogenicity of a single FVIII product in true PUPs. Inhibitor development was evaluated in 105 children treated for a median of 100 EDs. Inhibitors developed in 28 (26.7%) children after a median of 11 EDs. High-titer inhibitors [⩾5 Bethesda units (BU)/mL] developed in 17 (16.2%) children, after a median of 9 EDs, and low-titer inhibitors (⩾0.6 to <5 BU/mL) developed in 11 (10.5%) children after a median of 12 EDs. Five of the 11 low-titer inhibitors were transient and resolved without any change in the treatment regimen. No patients with non-null F8 mutations developed inhibitors. The cumulative incidence of inhibitors over time is shown in Figure 1. 36
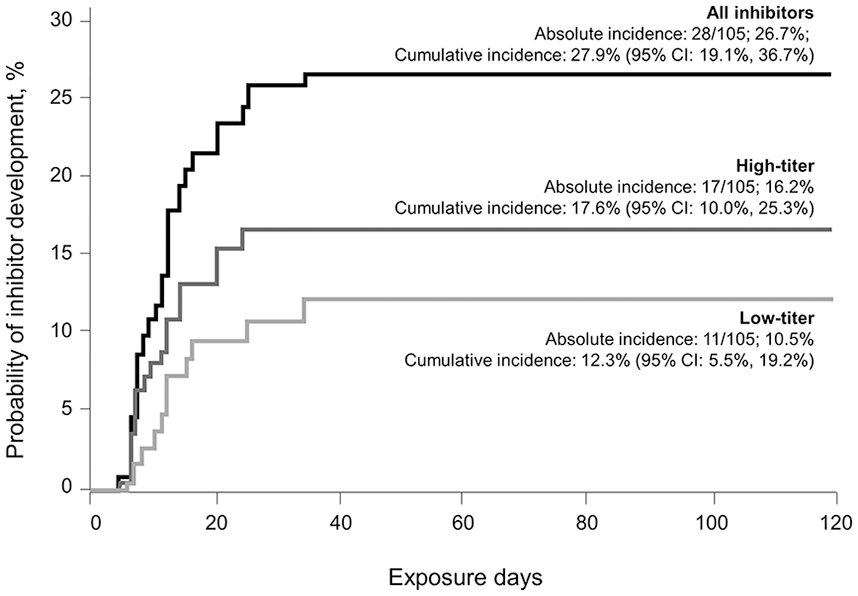
Incidence rates of inhibitor development in the NuProtect study. 36
The immunogenicity of other rFVIII products has been investigated in prospective trials in PUPs and minimally treated patients (MTPs), although differences in patient demographics and study design, such as differing follow-up times, make direct comparison challenging. Final reports from prospective studies with turoctocog alfa (NovoEight®) 40 and octocog alfa (Kovaltry®), 41 both rFVIII products manufactured in hamster cell lines, and with efmoroctocog alfa (Eloctate®), 42 a Fc-fusion rFVIII produced in a human cell line, have reported inhibitor incidence rates of 43.1%, 40 54.8%, 41 and 31.1%, 42 respectively. An inhibitor incidence rate of 30.0% was reported in PUPs treated with turoctocog alfa pegol (Esperoct®) a pegylated rFVIII product manufactured in a hamster cell line. 43 This study also reported that 17 PUPs experienced temporarily decreased incremental recovery, in the absence of FVIII inhibitors, in the first five EDs that were strongly correlated with anti-PEG antibody titers.43,44 In the prospective randomized SIPPET trial, the incidence of inhibitors was 37% in patients receiving a hamster cell line-derived rFVIII and 23% in those receiving a plasma-derived FVIII (pdFVIII). 45 The inhibitor incidence observed with simoctocog alfa, therefore, appears similar to that of pdFVIII in the SIPPET trial.45,46 Similarly, no inhibitors developed in patients with non-null F8 mutations in the pdFVIII arm of the SIPPET trial, as was the case in NuProtect, whereas the cumulative incidence of inhibitors in patients in the rFVIII arm of the SIPPET study was 47% for patients with null F8 mutations and 43% in patients with non-null F8 mutations. 46 Simoctocog alfa is produced in a human cell line and, therefore, has post-translational modifications similar to native human FVIII, whereas production of rFVIII in hamster cell lines results in post-translational modifications that differ from those found in pdFVIII (or simoctocog alfa). 29 Such differences have been suggested to affect the immunogenicity of the product. 29
Knowledge of the risk of inhibitor development with different FVIII products is needed to make informed decisions when treating PUPs. The ongoing non-interventional, observational Protect-NOW study (NCT03695978) will provide further insight into the immunogenicity of simoctocog alfa as well as the pdFVIII/VWF products (octanate® and wilate®). 47 The Protect-NOW study will follow PUPs in the real-world setting for up to 5 years (Figure 2). 47 Patients with prior emicizumab treatment are eligible for the Protect-NOW study, and concomitant use of emicizumab is permitted. 47

Protect-NOW study design. 47
Gaining and maintaining tolerance to FVIII is an important consideration in the management of patients with severe hemophilia A who receive emicizumab prophylaxis. 48 For patients on emicizumab who require surgery or treatment of breakthrough bleeds, if feasible, FVIII remains the safest and most effective approach for hemostatic management 49 ; however, if they have had limited or no exposure to FVIII, they are at risk of inhibitor development at any new exposure to FVIII which will occur with timing and modalities not comparable with the established experience of the pre-emicizumab era.39,48,50 Simultaneous administration of FVIII and emicizumab is a potential approach to achieve FVIII tolerance in PUPs that merits investigation.
The risk of inhibitor development in PTPs is low; however, cases have been reported with some FVIII products.39,51 In a recent study, inhibitors developed in 5 of 52 (9.6%) pediatric PTPs with severe or moderate hemophilia A after an institutional switch to moroctocog alfa (ReFacto AF®) in the Netherlands. 52 In earlier clinical trials with this product, an inhibitor rate of 1.5% (3 of 204) was reported in PTPs ⩾6 years old with moderate or severe hemophilia A. 53 In patients receiving turoctocog alfa pegol, one adult (0.6%) developed an inhibitor in a clinical trial of 175 PTPs aged ⩾12 years with severe hemophilia A. 54 No inhibitors developed to turoctocog alfa pegol in a clinical trial of 68 pediatric PTPs with severe hemophilia A. 55 In patients treated with octocog alfa (Advate®), one adult (0.4%) developed an inhibitor in clinical trials that included 276 pediatric and adult PTPs with moderate or severe hemophilia A. 56 No inhibitors have been reported in previously treated children who received simoctocog alfa during GENA-03, 32 GENA-13, 34 or the NuProtect-Extension 35 studies, or in clinical studies in adults PTPs.57,58 It is unknown whether tolerance to FVIII is maintained in PTPs who subsequently switch to emicizumab prophylaxis. Any loss of tolerance to FVIII in emicizumab-treated PTPs might increase the risk of inhibitor development if they require FVIII to treat acute bleeds or perioperatively or in case of switching to FVIII prophylaxis.
ITI with simoctocog alfa in children with inhibitors
The effectiveness of ITI with simoctocog alfa has been assessed in a retrospective case series of 10 children of various ethnicities with severe hemophilia A who had developed inhibitors to simoctocog alfa at three UK centers. 59 ITI success was defined as the achievement of at least two of three internationally recognized criteria: inhibitor titer <0.6 BU/mL, normalization of FVIII recovery (⩾66% of normal), and normalization of FVIII half-life (⩾6 h). Eight children had high-titer inhibitors (⩾5 BU/mL) and started ITI with 100 IU/kg daily (N = 7) or 50 IU/kg every other day (N = 1). Two children had low-titer inhibitors (<5 BU/mL) and started ITI on 90 and 100 IU/kg every other day. Negative inhibitor titers were achieved in eight (80%) patients in a median of 3.5 months, and ITI success was achieved by these eight (80%) children in a median of 7.5 months. 59
FVIII ITI with concomitant use of emicizumab is an attractive proposition that could provide improved bleeding control and reduce the intensity of FVIII dosing needed to eliminate inhibitors.18,49,60 The effectiveness and safety of simoctocog alfa ITI, either alone or in combination with emicizumab, is being assessed in an ongoing, 5-year, real-world study (MOTIVATE; NCT04023019 and EudraCT No. 2019-003427-38). 61
Prophylaxis with simoctocog alfa in children
Effective bleed prevention with prophylaxis offers children with severe hemophilia A the chance to live healthy and active lives, including participation in most physical and social activities. 6 Avoidance of repeated joint bleeds during childhood is also important to reduce the risk of hemophilic arthropathy, which impacts quality of life. 48
The efficacy and safety of simoctocog alfa prophylaxis have been demonstrated in both previously treated and untreated children in the simoctocog alfa clinical trial program. 31 In GENA-03, the median annualized bleeding rates (ABRs) for the 59 previously treated children were 0 for spontaneous bleeds and joint bleeds and 1.9 for all bleeds. 32 The long-term efficacy in bleed prevention was confirmed in 49 patients who continued into the GENA-13 study for a median of 30 months, with median ABRs of 0.3 for spontaneous bleeds, 0.4 for joint bleeds, and 1.7 for all bleeds. 34 In PUPs, 102 patients in the NuProtect study who received at least one prophylactic dose had median ABRs of 0 for spontaneous bleeds and joint bleeds and 2.8 for all bleeds during inhibitor-free periods. 33 These low rates were maintained in the 47 children who entered and were treated in the NuProtect-Extension study, with ABRs of 0 for spontaneous bleeds and joint bleeds and 1.0 for all bleeds. 35 During the median follow-up of 24 months, 34 (72%) patients had zero spontaneous bleeds. The most common initial prophylaxis regimen in the NuProtect-Extension was twice weekly (68%), followed by three times weekly (11%), every other day (6%), or once weekly (15%). At study completion, 85% of patients were on a twice-weekly regimen, 12% were treated three times weekly, and 3% were treated once weekly. 35 Analysis of efficacy over time in the NuProtect-Extension study demonstrated that 86–97% of children across all treatment regimens, and 90–97% of those on a twice-weekly regimen, had zero spontaneous bleeds (Figure 3). 35

Patients (%) with zero spontaneous bleeding events over time for any dosing regimen or a twice-weekly regimen in children receiving long-term simoctocog alfa prophylaxis in the NuProtect-Extension. 35
To further explore the long-term efficacy of simoctocog alfa prophylaxis in children, data were pooled from 96 children who took part in the GENA-05/15 (n = 47) and GENA-03/13 (n = 49) extension studies. 62 ABRs were calculated from the time of the patient’s first prophylactic dose in the main study to the completion of the extension study. The median age at prophylaxis start was 3 years. Children received simoctocog alfa prophylaxis for a median of 359 EDs and up to 5 years [median (range) 36.4 (10.9–59.2) months]. Median prophylactic doses were 35 IU/kg per ED and 96 IU/kg per week. 62 The dosing frequency in GENA-03/13 was three times weekly or every other day, whereas the dosing frequency in GENA-05/15 was at the discretion of the treating physician.32–35 The median ABRs for spontaneous and total bleeds over the entire prophylactic period were 0.3 and 1.8, respectively (Table 1). The median spontaneous and total joint ABRs were 0 and 0.4, respectively. Of the 96 children, 43 (45%) experienced no spontaneous breakthrough bleeds during a median duration of 36 months on prophylaxis. 62
ABRs during long-term simoctocog alfa prophylaxis in children (N = 96). 62

IQR, interquartile range.
The bleeding rates during long-term prophylaxis with simoctocog alfa34,35,62 are consistent with those reported from long-term extension studies investigating the efficacy of other rFVIII products, including extended half-life products, in children under 12 years of age.63–67 In these studies of other products, the median duration of prophylaxis ranged from 3.2 to 5.8 years, median spontaneous ABRs ranged from 0 to 0.6, and total ABRs from 0.8 to 2.0. In a 12-month study of once-weekly prophylaxis with efanesoctocog alfa in 74 children <12 years of age, the median ABR was 0 (interquartile range: 0–1). 68 The HAVEN-7 study is evaluating the efficacy and safety of emicizumab in infants (⩽1 year) with severe hemophilia A without FVIII inhibitors. 69 In an interim analysis of 54 PUPs/MTPs treated for a median (range) of 42 (1–60) weeks in HAVEN-7, the negative binomial regression model-based ABR for all bleeds was 1.9, and 43% of patients had zero bleeds. 69 In the context of these results, the data for simoctocog alfa in children provide insight into the bleeding tendencies of a large group of children receiving long-term prophylaxis with a single rFVIII product and demonstrates the efficacy of simoctocog alfa for long-term prophylaxis.
On-demand treatment of bleeds with simoctocog alfa in children
While prophylaxis is effective for the prevention of bleeds, children with hemophilia A may still experience breakthrough bleeds. Just as in healthy children, some of these bleeds are unavoidable, such as those caused by accidents, and the active nature of children may contribute to an increased frequency of traumatic bleeds in early years. 1 FVIII replacement therapy is the established and recommended approach for treatment of breakthrough bleeds during FVIII prophylaxis.6,70 In addition, FVIII replacement therapy is recommended for the treatment of breakthrough bleeds in patients on emicizumab prophylaxis in the absence of high-titer FVIII inhibitors.25–27 Quick and effective resolution of bleeds is an important focus in children to limit complications, prevent recurrence of bleeds, and reduce the risk of chronic joint disease and disability. 6
In the 96 children who received long-term simoctocog alfa prophylaxis, 86 children experienced a total of 851 breakthrough bleeds; 500 (58.8%) were minor, 342 (40.2%) were moderate to major, 5 (0.6%) were major to life-threatening, and 4 (0.5%) were of unknown severity. Of these, 763 breakthrough bleeds were treated with simoctocog alfa. One or two infusions were sufficient to resolve 86.5% of treated bleeds, and 85.1% of all bleeds were treated successfully, indicating that simoctocog alfa is effective in the treatment of breakthrough bleeds in children (Figure 4). 62

Breakthrough bleeds treated successfully (i.e. efficacy of treatment rated excellent or good by the patient/investigator) during long-term prophylaxis (N = 96). 62
The efficacy and safety of FVIII in treating breakthrough bleeds in non-inhibitor children on emicizumab prophylaxis have not been specifically studied in clinical trials. However, post hoc data from a clinical trial in patients ⩾12 years of age 71 and real-world data in children 72 have demonstrated the efficacy of FVIII in the treatment of breakthrough bleeds in non-inhibitor children on emicizumab prophylaxis.
Perioperative use of simoctocog alfa in children
Hemophilia A patients are at risk of bleeding complications both during and after surgery.6,73 Perioperative administration of FVIII remains the established approach to maintaining hemostasis, allowing hemophilia A patients to undergo surgeries with a bleed risk similar to that of the general population.73,74 However, surgeries are performed less frequently in children with hemophilia A than in adults, resulting in relatively limited information for this group of patients. 75
During the clinical trial program, simoctocog alfa was used as surgical prophylaxis for 56 surgeries in children, of which 27 were major surgeries.32,34 The hemostatic efficacy was rated as ‘excellent’ or ‘good’ for 42 of 43 (98%) surgeries (15 of 16 minor and 27 of 27 major) with ratings available from the surgeon and hematologist. These data demonstrate that simoctocog alfa successfully maintained hemostasis and in children with severe hemophilia A.
Additional FVIII treatment is often required in non-inhibitor patients on emicizumab undergoing surgery.25–27 Post hoc data from a clinical trial in patients ⩾12 years of age 76 and real-world studies in children77,78 have shown that perioperative FVIII is effective in emicizumab-treated children without inhibitors. An ongoing phase IV study (NuPOWER; NCT05935358) is assessing perioperative simoctocog alfa in patients aged ⩾12 years of age who are receiving emicizumab prophylaxis and undergoing major surgery. 79
Safety of simoctocog alfa in children
The safety of simoctocog alfa was evaluated in all 167 children who participated in the GENA clinical trials, with patients receiving a total of 43,773 infusions throughout all four studies. In addition to inhibitor development in PUPs in NuProtect, treatment-related adverse events reported across all four studies were pyrexia [19 patients (1 patient in GENA-13 and 18 patients in NuProtect)], hypersensitivity (8 patients in NuProtect), back pain (1 patient in GENA-03), headache (1 patient in GENA-03), dyspnea (1 patient in GENA-13), anemia (1 patient in NuProtect), and hemorrhagic anemia (1 patient in NuProtect). Pyrexia was classified as serious in three patients (one in GENA-13 and two in NuProtect), and hypersensitivity (rash) was classified as serious in one patient in NuProtect. No thromboembolisms were recorded and there were no deaths related to treatment in any of the trials.
Bone and joint health
Hemophilic arthropathy develops due to repeated bleeds into the joint and is a significant cause of morbidity in people with hemophilia A. 5 Avoiding its development is important, especially in children, to prevent long-term disability, pain, and negative impact on quality of life. 6 Early initiation of FVIII prophylaxis has been shown to provide protection against joint damage.80,81 In hemophilia A, clinical assessment of joint structure and function is commonly performed using the Hemophilia Joint Health Score (HJHS) in children and adolescents. 6 In the simoctocog alfa clinical trial program, HJHS was used to assess joint health in previously treated children in GENA-03 and GENA-13. 74 In those children who took part in GENA-13 for whom HJHS values are available, the median HJHS was 0 (mean 0.6) at enrollment into GENA-03 and 0 (mean 0.2) at the end of GENA-13. 62 A number of clinical and scientific studies are ongoing to investigate in more detail the potential impact of simoctocog alfa on bone and joint health.
Some patients develop joint damage despite a lack of clinically evident joint bleeds.5,80 Subclinical changes in joint structure have been observed by magnetic resonance imaging (MRI) in hemophilia patients without clinically evident bleeds in those joints.5,80 Although the sensitivity of joints to even a single clinically identified bleed has been demonstrated,82,83 it remains unknown to what degree subclinical bleeds may impact long-term joint health. Therefore, detection of early signs predictive of joint damage may help optimize management approaches in patients with hemophilia A. An optional sub-study of the ongoing MOTIVATE study is evaluating levels of established bone and joint health biomarkers, as well as aiming to identify and validate new markers of early-onset joint degeneration. 61 A 4-year prospective study (PROVE) is planned to compare the long-term effects of simoctocog alfa and emicizumab on joint health in routine clinical practice. The PROVE study will enroll patients ⩾12 years of age and will compare joint health using MRI (primary endpoint), HJHS, and bone and joint biomarkers.
Differences in bone mineral density (BMD) are observed from an early age in children with hemophilia A compared with the general population84,85; FVIII prophylaxis has been shown to partially ameliorate this difference. 86 However, the underlying molecular mechanisms and role of FVIII in maintaining BMD remain poorly understood and is an area of growing interest. 87 In vitro studies are ongoing to assess this, as well as the impact of simoctocog alfa on bone metabolism. The PROVE study will measure changes in BMD over 4 years in patients ⩾12 years of age treated with simoctocog alfa or emicizumab.
Conclusion
Despite the rich treatment landscape of hemophilia A, which now includes FVIII mimetics, antagonists to natural coagulation inhibitors, and gene therapy, FVIII replacement remains an indispensable treatment option for patients, including children, with hemophilia A. Coagulation activation provided by non-factor therapies and levels of FVIII expression with gene therapy are not always adequate to face all the clinical challenges. Furthermore, at this stage, gene therapy is not licensed for children. In this light, it is important to have effective FVIII replacement to rely upon in case of bleeding and/or surgery/trauma. In this respect, achieving tolerance toward FVIII remains relevant in the management of hemophilia A. Comprehensive data, including data on long-term prophylaxis for up to 5 years, show that simoctocog alfa is efficacious and well tolerated for the prevention and treatment of bleeds and for surgical coverage in children with hemophilia A. A low rate of inhibitor development was observed in previously untreated children receiving simoctocog alfa, and further data on the immunogenicity of simoctocog alfa are being collected in the real-world Protect-NOW study. Real-world data have also demonstrated the effectiveness of ITI with simoctocog alfa in children with inhibitors. Finally, the ongoing studies aim to gain greater insight into how simoctocog alfa may address the remaining unmet needs of children with hemophilia A and help to improve long-term outcomes.