
Abstract
Post-translational modifications of the nucleosomal histone proteins orchestrate chromatin organization and gene expression in normal and cancer cells. Among them, the acetylation of N-terminal histone tails represents the fundamental epigenetic mark of open structure chromatin and active gene transcription. The bromodomain and extra-terminal (BET) proteins are epigenetic readers which utilize tandem bromodomains (BRD) modules to recognize and dock themselves on the acetylated lysine tails. The BET proteins act as scaffolds for the recruitment of transcription factors and chromatin organizers required in transcription initiation and elongation. The recent discovery of small molecules capable of blocking their lysine-binding pocket is the first paradigm of successful pharmacological inhibition of epigenetic readers. JQ1 is a prototype benzodiazepine molecule and a specific BET inhibitor with antineoplastic activity both in solid tumours and haematological malignancies. The quinolone I-BET151 and the suitable for clinical development I-BET762 benzodiazepine were introduced in parallel with JQ1 and have also shown potent antitumour activity in preclinical studies. I-BET762 is currently being tested in early phase clinical trials, along with a rapidly growing list of other BET inhibitors. Unlike older epigenetic therapies, the study of BET inhibitors has offered substantial, context-specific, mechanistic insights of their antitumour activity, which will facilitate optimal therapeutic targeting in future. Here, we review the development of this novel class of epigenetic drugs, the biology of BET protein inhibition, the emerging evidence from preclinical work and early phase clinical studies and we discuss their potential role in the treatment of haematological malignancies.
Keywords
Introduction
The importance of epigenetic gene expression deregulation, in addition to genetic aberrations, has been increasingly recognized in the pathogenesis of haematological malignancies. In fact, a number of ‘epigenetic’ therapies targeting DNA methylation and posttranslational histone modi-fications have been already introduced in clinical practice [Campbell and Tummino, 2014]. Aberrant DNA hypomethylation and hypermethylation patterns, linked with gene overexpression or silencing respectively, have been described in myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML) [Figueroa et al. 2009; Jiang et al. 2009]. In addition, recurrent genetic events involving epigenetic regulators have been identified in AML with typical examples being mutations in the DNA methyltransferase 3a (DNMT3a) gene and chromosomal rearrangements involving the mixed lineage leukaemia (MLL) gene [Gilliland et al. 2004; Krivtsov and Armstrong, 2007; Ley et al. 2010]. The DNA methyltransferase inhibitors azycitidine and decitabine are approved for clinical use in MDS [Silverman, 2004; Silverman and Mufti, 2005; Raj and Mufti, 2006]. As well as DNA methylation, epigenetic therapeutic targeting has also focused on epigenetic ‘writers’ (e.g. histone acetyl-transferases) and ‘erasers’ (e.g. histone deacetylases [HDAC]), proteins with enzymatic activity that either add or remove post-translational modifications respectively to histone proteins. Vorinostat is the first HDAC inhibitor approved for treatment of relapsed cutaneous T-cell lymphoma [Duvic et al. 2007; Grant et al. 2007; Mann et al. 2007] and along with other HDAC inhibitors such as romidepsin and panobinostat, is in clinical trials for other types of lymphoma and for multiple myeloma [Dimopoulos et al. 2013; Coiffier et al. 2014; Straus et al. 2014].
However, despite some successful clinical paradigms, lack of consistent clinical efficacy, off-target effects and toxicity and the incomplete understanding of mechanisms of action, have raised scepticism around the concept of ‘epigenetic’ therapy [Grant, 2009; Griffiths and Gore, 2013; Treppendahl et al. 2014].
The recent development of small molecule inhibitors of the bromodomain and extra-terminal domain (BET) family of proteins represents a new chapter in ‘epigenetic therapy’ not just because they are the first example of successful pharmacological interference with epigenetic ‘readers’, i.e. proteins that read histone post-translational modifications. BET protein inhibitors are also a fine example of development of highly selective designer drugs guided by high quality structural and functional data. Preclinical work with BET protein inhibitors, facilitated by modern advances in next-generation sequencing (NGS) and genome-wide technologies, has generated a wealth of context-specific mechanistic data in various types of haematological malignancies. As a result, only 4 years after their first description, several inhibitors are already in early clinical development and some encouraging preliminary results have been reported.
In this review, we discuss the biological function of the BET protein epigenetic ‘readers’ and the rationale for their pharmacological inhibition. We present exciting mechanistic knowledge that emerged from the use of BET protein inhibitors alongside convincing preclinical evidence of antitumour activity and how this new knowledge sets the basis for the clinical development of BET inhibitors in haematological malignancies.
Histone acetylation and bromodomains
Allfrey and colleagues described lysine residue acetylation, the first post-translational histone modification, in 1964 and they also proposed its functional importance [Allfrey et al. 1964]. Acetylation is the most prominent chromatin modification. It is generally linked with open structure chromatin and active gene transcription. It is a dynamic process, tightly regulated by two enzyme groups with opposing effects, the histone acetyl-transferases (HAT) and the HDAC. HAT perform the enzymatic transfer of an acetyl group from the acetylCoA to the ε-amino group of lysine side chains of histone protein tails and display target selectivity [Shahbazian and Grunstein, 2007]. Lysine acetylation changes the electrostatic interaction between histones or histones and DNA and allows higher accessibility to DNA by changing the nucleosome positioning and chromatin architecture. More importantly, lysine acetylation provides an instructive pattern for the recruitment to chromatin of factors involved in transcription regulation, DNA repair and replication [Kouzarides, 2007; Shahbazian and Grunstein, 2007].
Bromodomains (BRD) are the only protein interaction modules that preferentially bind ε-N-acetylated lysine residues through structurally well-defined pockets. In humans, there are 8 BRD-containing protein families comprising 46 nuclear or cytoplasmic proteins with diverse structures and functions.
Differences in the acetyl-binding pockets of BRD are important not only for the characterization of the BRD proteins but also for design of selective BRD inhibitors [Dhalluin et al. 1999; Filippakopoulos et al. 2012b]. The BRD proteins include, among other members, chromatin-modifying enzymes such as HAT and HAT-associated proteins (GCN, CREBBP and EP300), methyltransferases (MLL, ASH1L), helicases (SMARCA2 and SMARCA4), chromatin remodellers (BAZ1A and BAZ1B), transcriptional co-activators and mediators (TAF1, SP100, SP110), and the BET family [Filippakopoulos and Knapp S., 2012; Filippakopoulos et al. 2012b].
The BET family consists of BRD2, BRD3, BRD4 and BRDT. Expression of BRD2–4 is ubiquitous, while BRDT expression is restricted to germ cells. BET proteins contain two conserved N-terminal BRD modules (BD1 and BD2) able to specifically recognize and bind acetylated lysine residues, but also to interact with and function as scaffolds for a number of other molecules implicated in gene transcription. Although the advent of BET inhibitors has tremendously advanced our understanding of BET protein biology, the rationale for therapeutic targeting of BET proteins was based on preceding pivotal functional studies. Early work showed that Brd2 is specifically recruited to acetylated histones H3 and H4 and this interaction is linked to active transcription and mitosis [Kanno et al. 2004; Huang et al. 2007]. Brd2 and Brd3 are required for permissive RNA polymerase II (Pol II) transcription through acetylated nucleosomes [Leroy et al. 2008]. In eukaryotic cells, Brd4 binds acetylated histones using both BRD modules. The BD2 domain also recognizes and interacts with the acetylated region of cyclin T1 [Schroder et al. 2012]. Cyclin T1 and Cdk9 comprise the heterodimeric complex of the active form of the positive transcription elongation factor b (P-TEFb). In addition, Brd4 controls the release of active P-TEFb from its inactive complex with HEXIM1 protein and 7S snRNA. The recruitment of active P-TEFb is crucial for the sustained presence of Pol II in active genes and for transcription initiation and elongation. The recruitment of P-TEFb and Pol II by Brd4 is crucial for the expression of cell proliferation supporting genes, including c-Myc and its target genes [Rahl et al. 2010]. Brd4 is a key regulatory factor for the expression of genes required in M to early G1 phase transition [Yang et al. 2005, 2008].
In the pre-BET inhibitor era, research on the role of Brd4 in cancer biology was hampered by the fact that Brd4 deletion in mice was embryonic lethal. [Houzelstein et al. 2002]. Nevertheless, Brd4 inhibition by short hairpin RNA (shRNA) had shown antileukaemic potential in AML [Zuber et al. 2011] while Brd2 overexpression in lymphocytes of transgenic mice results in the development of splenic B cell lymphomas, with histological features and transcriptome profiling similar to human diffuse large B cell lymphomas (DLBCL) [Greenwald et al. 2004].
Only few genetic events have linked BET proteins with cancer pathogenesis [French et al. 2003; Crawford et al. 2008 ]. The most prominent human cancer paradigm is the NUT midline carcinoma (NMC), a rare but aggressive form of intrathoracic squamous cell carcinoma. In most cases, the disease is characterized by a single genetic aberration involving the nuclear protein of testis (NUT) gene, either in the balanced translocation t(15;19)(q14;p13.1), which generates the BRD4-NUT fusion gene or in a similar translocation involving BRD3 [French et al. 2001; French et al. 2008]. The BRD4-NUT fusion protein blocks differentiation and supports a high cell proliferation rate. siRNA knockdown of BRD4-NUT irreversibly lifts the differentiation blockage and induces proliferation arrest in NMC cell lines [French et al. 2008]. Some mechanistic aspects of the BRD4-NUT malignant transformation in NMC have been unfolded in recent studies. BRD4-NUT and BRD4 colocalize in acetylated but nontranscribed chromatin sites. The NUT moiety of the fusion protein recruits HAT, such as p300, resulting in hyperacetylation and further sequestration of BRD4, along with P-TEFb and other transcription machinery components. This functional depletion of BRD4 leads to transcriptional repression of c-fos, a critical gene for epithelial differentiation. In addition, p300 sequestration in inactive chromatin domains results in p53 inactivation. BRD4-NUT knockdown restores c-fos expression and p53 activity [Reynoird et al. 2010; Yan et al. 2011].
The development of the BET proteins inhibitors
Early attempts to identify small molecule BRD inhibitors were based on the structural characterization of the BRD acetyl-binding pocket and nuclear magnetic resonance spectroscopy screening of a large number of candidate compounds [Dhalluin et al. 1999; Sachchidanand et al. 2006]. Notably, these studies did not focus on BET proteins, but the acetyltrasferase CREB-binding protein (CBP). CBP acetylates and with its BRD binds the acetyl-lysine 382 of the p53 tumour suppressor protein to modulate p53 stability and function in DNA damage repair [Sachchidanand et al. 2006; Borah et al. 2011]. These studies identified chemical compounds with micromolar level affinity for the BRD pocket and therefore unsuitable for clinical development; nevertheless, they showed that in principle BRD inhibition was feasible [Borah et al. 2011].
In 2010, two potent and importantly, selective BET protein inhibitors were reported. The first inhibitor, JQ1, a thieno-triazolo-1,4-diazepine, was based on a previous observation that some novel benzodiazepines inhibit BET proteins [Filippakopoulos et al. 2010; Adachi et al. 2011; Miyoshi et al. 2013]. In fact, some of the benzodiazepines already in clinical use also bind BRD4, but with low affinity [Straw, 1985; Filippakopoulos et al. 2012a]. JQ1 displaces BRD4 from nuclear chromatin at nanomolar concentrations and was active against NUT midline carcinoma cell lines in vitro and in a relevant xenograft model, thus establishing one of the first paradigms of successful BET inhibition in oncology [Filippakopoulos et al. 2010].
In parallel, a GlaxoSmithKline group identified I-BET762, also known as GSK525762A, a novel benzodiazepine which binds selectively to the acetyl-recognising BET pocket with nanomolar affinity [Nicodeme et al. 2010; Gosmini et al. 2014].
Since BET proteins are required for active transcription initiation and elongation of inflammatory genes [Hargreaves et al. 2009] I-BET762 was used in an inflammatory disease model. I-BET762 potently inhibited expression of lipopolysaccharide (LPS)-inducible genes in macrophages in vitro and it suppressed inflammation in a mouse model of severe sepsis [Nicodeme et al. 2010]. Preclinical evidence of I-BET762 antitumour activity in myeloma, acute leukaemia and solid cancers, including the NUT midline carcinoma was demonstrated in later studies. Owing to its favourable pharmacological profile I-BET762 is one of the several BET protein inhibitors currently tested in early phase clinical trials [Mirguet et al. 2013; Wyce et al. 2013a; Zhao et al. 2013b; Asangani et al. 2014; Chaidos et al. 2014].
The exciting results from JQ1 and I-BET762 and the favourable pharmacological properties of the diazepine chemical template encouraged the development of similar structure BET inhibitors. A list of benzodiazepine BET inhibitors and relevant clinical trials are summarized in Table 1 [Smith et al. 2014]. MS417 shares the same thieno-triazolo-1,4-diazepine scaffold with JQ1. In the HIV-1 Tg26 transgenic mice, MS417 blocks BRD-4 binding to acetylated NF-κB and effectively attenuates the inflammatory response in HIV-induced nephropathy [Zhang et al. 2012a]. MS417 induces cell cycle arrest in preclinical melanoma models, but it has not been tested in haematological malignancies [Paoluzzi et al. 2013]. OTX015, originally developed for the treatment of inflammatory bowel disease, is among the thienodiazepine inhibitors with a promising antitumour profile in haematological malignancies [Miyoshi et al. 2009; Gautschi and Minikis, 2014]. In a phase I acute leukaemia study, OTX015 induced remissions, including complete remission in two patients with refractory disease [Boi et al. 2012; Bonetti et al. 2012a, 2012b; Braun et al. 2013; Noel et al. 2013]. CPI-203 is another benzodiazepine inhibitor structurally similar to JQ1, but with improved bioavailability profile in mice [King et al. 2013]. It has shown preclinical evidence of efficacy in lymphomas [Ceribelli et al. 2014; Moros et al. 2014].
BET protein inhibitors in early phase clinical trials.

I-BET151 (GSK1210151A), is a highly specific and potent isoxazoloquinoline BET inhibitor, with a pharmacokinetic and bioavailability profile compatible for future clinical development [Dawson et al. 2011; Hewings et al. 2011; Mirguet et al. 2012; Seal et al. 2012]. In preclinical setting, I-BET151 is an effective inhibitor of interleukin 6 production by LPS-stimulated peripheral blood mononuclear cells (PMBC) [Seal et al. 2012] and has antitumour activity in haematological malignancies including myeloma [Chaidos et al. 2014], acute myeloid leukaemia [Dawson et al. 2011; Dawson et al. 2014], lymphoma [Tolani et al. 2014] and myeloproliferative neoplasms [Wyspianska et al. 2014]. More recently, I-BET151 showed effective inhibition of osteoclastogenesis and inflammatory bone resorption [Park-Min et al. 2014].
RVX-208 is a quinazolone derivative of resveratrol and it has been tested already in hundreds of patients in phase II clinical trials for the treatment of atherosclerosis. Interestingly, unlike other BET inhibitors, RVX-208 binds preferentially to the BD2 domain of BRD2 and BRD3 proteins. RVX-208 provides proof-of-concept that selective inhibition within the BET family is feasible [Picaud et al. 2013; Jahagirdar et al. 2014]. This observation may have clinical implications in cancer, for instance the development of BET inhibitors that spare BRDT and therefore may be more suitable for the paediatric population.
RVX-2135 is based on the same chemical scaffold as RVX-208. Although it preferentially binds BD2, it is a pan-BET protein inhibitor, with anti-tumour activity in AML and murine lymphoma [Campeau et al. 2013; Bhadury et al. 2014].
Mechanistic aspects of context-specific BET inhibition in haematological malignancies
The discovery of potent BET bromodomain inhibitors and their therapeutic activity in inflammation and cancer has offered unprecedented opportunity to gain mechanistic insights into the role of BET proteins in transcriptional regulation and disease biology in general. The engagement with BET inhibitors dislocates BRD4 from the histone acetyl marks, but in addition disrupts the interaction with Cyclin T1 and hence with P-TEFb, allowing its natural sequesters HEXIM1/7S snRNA to retain P-TEFb bound in an inactive complex [Jang et al. 2005; Yang et al. 2005; Chen et al. 2014].
BRD4 is a global regulator of gene transcription, therefore it would be expected that BRD4 inhibition would impact on the transcription activity across all active genes. In fact, there is only a modest reduction in global gene expression and mRNA levels. BRD4 inhibition leads significant reduction of the transcript levels of only a few hundreds genes, interestingly in a cell- and context-specific manner. In a characteristic example, Delmore and colleagues showed that JQ1 treatment of the myeloma cell line MM1.S induces drastic transcriptional downregulation of the MYC gene and MYC-dependent genes, while other transcription factors that are important for the biology of myeloma are not affected [Delmore et al. 2011].
The molecular basis of the transcriptional selectivity exhibited by BET inhibitors was delineated in an elegant study by Loven and colleagues [Loven et al. 2013]. This work was based on previous observations of preferential BRD4 occupancy in enhancers, at much higher levels than at the promoters of active genes [Delmore et al. 2011]. ChIP-seq analysis in MM1.S cells was used to chart genome-wide occupancy of BRD4 and MED1 (a Mediator subunit). Coinciding with RNA Pol II, signals of BRD4 and MED1 were found at transcription initiation sites, but, additionally, BRD4, together with the enhancer-associated H3K27Ac mark, was found enriched in about 8000 regions far beyond transcription initiation sites. Further analysis, identified 308 genomic areas within a 50 kb window from the transcription initiation sites, with a remarkable 16-fold and 18-fold higher occupancy by BRD4 and MED1 respectively compared with typical enhancers. These 308 regions were termed super-enhancers and occupy about 40% of the total enhancer-bound BRD4 and MED1. Super-enhancers in MM1.S cells are associated with key genes in myeloma biology, characterized by high expression levels, including MYC, IRF4, PRDM1 and XBP1 [Loven et al. 2013]. The asymmetric BRD4 occupancy in a limited number of super-enhancers was confirmed in a DLBCL study, showing that 33% of the total BRD4 localizes at super-enhancers which are predicted to regulate just 1.6% of all actives genes. Super-enhancers are associated with key transcription factors implicated in lymphomagenesis, including MYC, BCL6, IRF8, PAX5, OCA-B [Chapuy et al. 2013]. The super-enhancers have key roles in healthy cells, haematological and solid tumours [Carro et al. 2010; Borggrefe and Yue, 2011; Conaway and Conaway, 2011; Hnisz et al. 2013]. The oncogenic transcription supported by super-enhancers is extremely sensitive to BRD4 inhibition. For instance, treatment of the MM1.S myeloma cells with low concentrations of JQ1 has no impact on global mRNA levels but it results in tremendous depletion of MYC and IRF4 mRNA [Loven et al. 2013]. These data offer crucial mechanistic insights on the selective anti-tumour activity of BET inhibitors, but also provide a valuable background for the rationalized design of future clinical studies (Figure 1).
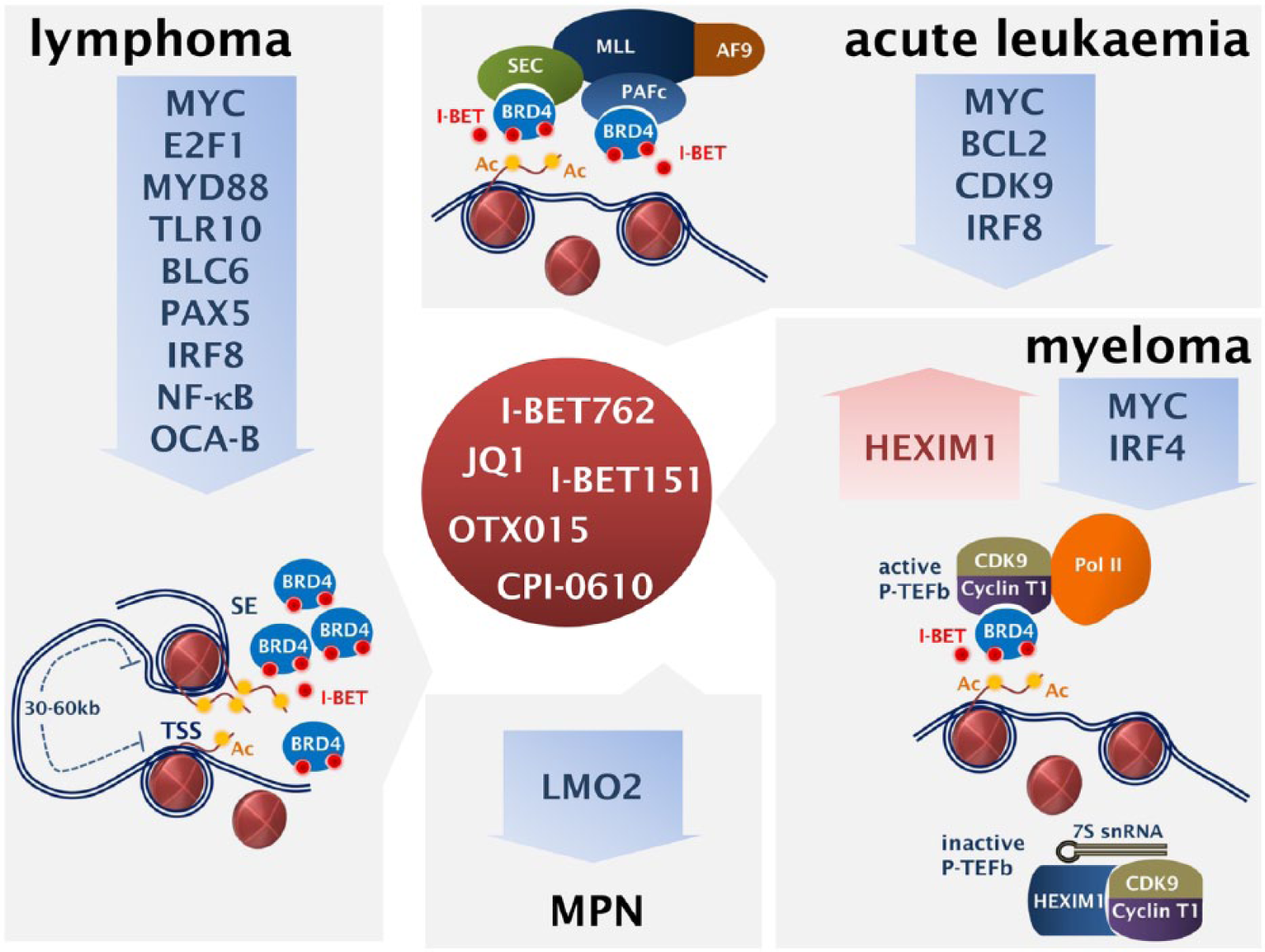
Mechanisms of action of the bromodomain and extra-terminal (BET) protein inhibitors: main genes and gene signatures targeted in acute leukaemia, myeloma, lymphoma and myeloproliferative neoplasms (MPN) are shown. SE, super-enhancer; TSS, transcription starting sites.
The emerging preclinical and clinical evidence in haematological malignancies
Acute leukaemia
Recent advances in the molecular understanding of leukaemogenesis including the discovery of recurrent genetic events that affect chromatin regulators, emphasize the importance of epigenetic deregulation in gene transcription in acute leukaemia. A classic example of epigenetic deregulation is the MLL-fusion leukaemia, which comprises >70% of paediatric acute lymphoblastic leukaemia (ALL) cases and 10% of adult AML. MLL is a histone methyltransferase responsible for ‘writing’ the histone activating mark H3K4me3. Leukaemia-associated translocations lead to rearrangement of MLL, a chromosome 11q23 gene, and its fusion with over 50 different partners. Many of these partners are critical components of the initiation and transcription elongation complexes SEC and PAFc [Krivtsov and Armstrong, 2007]. Two research groups used the MLL-fusion leukaemia model to produce one of the first successful paradigms of therapeutic BET inhibition in haematology.
Dawson and colleagues applied a global proteomics discovery approach to precipitate BRD proteins and associated protein complexes from nuclear extracts on beads coated with either I-BET762 or acetylated H4. They showed that BET proteins co-immunoprecipitate with components of the SEC and PAFc complexes [Dawson et al. 2011], which are involved in transcription elongation and have crucial roles in the malignant transformation of MLL-fusion leukaemia [Lin et al. 2010; Yokoyama et al. 2010]. This observation provided the rational for BET inhibition aiming to displace SEC and PAFc from chromatin and induce a therapeutic effect. Human MLL-fusion driven leukaemia cell lines and retrovirally transformed murine progenitors were extremely sensitive to nM concentrations of I-BET151, which induced rapid induction of cell cycle arrest, apoptosis and complete ablation of clonogenic potential. Similar effects were observed in primary human blasts, from patients with MLL-fusion leukaemias. Remarkably, transcriptome analysis revealed downregulation of genes and gene signatures that overlap with the MLL oncogenic programme and its direct targets, including MYC, CDK6 and BCL2. In a xenotransplantation model of established, disseminated human MLL-AF4 leukaemia daily I-BET151 injections achieved excellent disease control and significant survival benefit [Dawson et al. 2011].
In a different global approach aiming to delineate the epigenetic deregulation in AML, Zuber and colleagues used a custom library of 1094 shRNA to target 243 known chromatin regulators and placed the shRNA in a doxycycline-induced promoter to avoid bias. shRNA screening in the MLL-AF9/NrasG12D murine AML cell line identified five independent shRNAs with Brd4 knowndown effect, leading to cell cycle arrest and apoptosis. In an established murine AML model, the Brd4 shRNAs controlled disease progression and conferred survival advantage. JQ1 effectively induced cell proliferation arrest and apoptosis in most MLL-fusion and non-MLL acute leukaemia cell lines and primary samples tested. The authors suggested that downregulation of Myc and Myc-driven gene signatures specific to the leukaemia stem cell population mediate the JQ1 therapeutic effects in AML [Zuber et al. 2011].
More recently, the antitumour activity of I-BET151 across different AML subtypes, including those associated with NPM1c mutation, was conformed [Dawson et al. 2014]. Wild-type nuclear NPM1 protein colocalizes with BRD4 and represses its transcriptional functions. The NPM1c mutation lifts this repressive effect and upregulates a transcriptional programme, which increases sensitivity to I-BET151, both in vitro and in the NOD SCID transplanted mice [Dawson et al. 2014].
Taken together, these data suggest that BET inhibitors are a promising therapeutic approach across all genetic subtypes of AML. MYC downregulation appears as a unifying mechanistic theme, but other mechanisms of action shown in MLL-fusion and NPM1c mutated AML could be harnessed to further facilitate optimal therapeutic targeting.
Compelling preclinical evidence of BET inhibitors antileukaemic activity both in B- and T-cell ALL has also emerged from a number of studies. In B ALL, a clear antiproliferative and pro-apoptotic effect was seen with JQ1 treatment in vitro and in xenotransplantation models. Along with MYC, an equally strong depletion of interleukin 7 receptor gene (IL7R) transcripts was observed, confirmed by reduced IL7R surface expression. IL7R expression is restricted to early lymphoid lineage cells and plays an important role in normal development and leukaemogenesis [Ott et al. 2012]. JQ1 downregulates MYC and induces cell cycle arrest and apoptosis in T lymphoblastic cell lines and primary cells, including treatment-refractory paediatric T-ALL, both in vitro and in NSG transplanted mice. JQ1 is effective regardless of the underlying oncogenic events, including Notch-driven ALL, refractory to Notch inhibitors [Roderick et al. 2014]. The latter was linked to a distal Notch1 enhancer which ordinarily interacts with the Myc proximal promoter. However, in Notch inhibitor-refractory cells, Myc transcription is driven by an alternative, Brd4-dependent promoter, hence the sensitivity of these cells to Brd4 inhibition. This is an interesting example of overcoming drug resistance with BET protein inhibitors and provides the rationale for combination therapies in T ALL [Yashiro-Ohtani et al. 2014].
I-BET762, OTX015 and CPI-0610 are currently being evaluated in phase I clinical trials (Table 1). Preliminary results have been reported from the OTX015 trial in refractory haematological malignancies. A total of four of 14 patients with relapsed/refractory secondary or therapy-related AML receiving OTX015 at either 80 mg once daily or 40 mg twice daily had measurable responses, including one complete remission (CR) and one CR with incomplete blood count recovery [Herait et al. 2013]. This is the first published evidence of clinical activity for a BET protein inhibitor in patients with advanced haematological malignancy.
Multiple myeloma
The preclinical evidence of antimyeloma activity of BET protein inhibition was first demonstrated with JQ1 treatment of myeloma cell lines, in xenograft myeloma models and the Vk*MYC transgenic mice. JQ1 induces cell proliferation arrest in myeloma cell lines (IC-50 68–502 nM) and pronounced cell senescence, even in the presence of HS-5 stromal cells. A similar effect was seen in CD138+ primary cells from patients with relapsed/refractory myeloma. The in vivo therapeutic potential of JQ1 was tested in an orthotopic mouse xenograft model using MM1.S-luc cells. The animals were given daily intraperitoneal JQ1 injections, resulting in significant reduction of myeloma tumour burden and prolonged survival. In the Vk*MYC mice, JQ1 caused significant reduction of the serum M-spike concentration, including complete remission in one animal. It should be noted that the JQ1 activity in this study was tested solely in the context of MYC genetic aberrations and it was directly attributed to MYC downregulation [Delmore et al. 2011].
Another study, aiming to validate Vk*MYC as a reliable murine model of myeloma for drug screening, compared several single agents with established or unknown therapeutic profile in myeloma. JQ1 induced a remarkable, >50% M-spike reduction, similar to standard antimyeloma agents including alkylating agents, bortezomib and dexamethasone [Chesi et al. 2012].
We studied the antimyeloma potential of I-BET151 and I-BET762 in vitro and in xenograft assays. Both inhibitors induce cell cycle arrest (IC50 100–300 nM) and apoptosis in all myeloma cell lines tested, in a time- and dose-dependent manner, in stroma and nonstroma conditions. Of note, the cell lines were not selected for their cytogenetic profile, including the presence of MYC aberrations. I-BET151 is also active against primary CD138+ myeloma cells in vitro, and overcomes the stroma and interleukin 6-mediated antiapoptotic effects. Both inhibitors were active in vivo in subcutaneous and systemic myeloma xenograft models. Interestingly, transcriptome analysis of H929 and KMS12BM cells after exposure to I-BET151 showed little overlap in downregulated genes between the two cell lines. This finding may reflect differences in the cytogenetic profiles and further emphasizes the selectivity of BET protein inhibition. As well as downregulation of MYC and MYC-dependent signatures, particularly in KMS12BM cells, downregulation of IRF4-dependent oncogenic signatures and of other myeloma-specific transcriptional programmes were also observed. In keeping with the same notion of MYC-independent mechanisms of antitumour activity, we showed that HEXIM1 transcripts and protein levels increase with I-BET151 treatment [Chaidos et al. 2014]. HEXIM1 upregulation is a consistent observation in GEP performed after BET inhibition in other malignancies, including lymphoma and neuroblastoma [Delmore et al. 2011; Mertz et al. 2011; Chapuy et al. 2013; Puissant et al. 2013]. HEXIM1 sequesters and reduces the availability of CDK9, the catalytically active component of P-TEFb, required for PolII activation. Therefore, depletion of active P-TEFb by HEXIM1 has an additional negative impact on actively transcribed genes which is separate from the direct inhibition of BRD4 binding to chromatin. Our data suggest that HEXIM1 upregulation in response to BET protein inhibitors in myeloma is not BRD4 mediated, however it leads to reduced active P-TEFb availability and contributes to the biological effects of BET inhibitors [Chaidos et al. 2014]. Such a role for HEXIM1 in transcriptional regulation has been also suggested elsewhere. HEXIM1 is a P-TEFb dependant gene [He et al. 2006]. A temporary release of P-TEFb from its sequesters, induces HEXIM1 upregulation, leading to perturbation of the P-TEFb equilibrium between active and inactive forms. HEXIM1 upregulation induced growth arrest in tumours and defines transcriptional programmes in pluripotent cells [He et al. 2006; Bartholomeeusen et al. 2012; Liu et al. 2014].
Lymphoma
The first evidence of BET-mediated deregulation of gene expression in lymphomagenesis was demonstrated with the development of aggressive B-cell lymphomas in transgenic mice with lymphoid-restricted Brd2 overexpression [Greenwald et al. 2004]. On the other hand, studies in myeloma, MLL-fusion leukaemia and neuroblastoma very clearly had showed that BET inhibition downregulates critical genes for lymphoma biology, including MYC, IRF4 and BCL2 and provided a rational basis for the study of BET protein inhibition in lymphoma [Dawson et al. 2011; Delmore et al. 2011; Mertz et al. 2011; Wyce et al. 2013b].
In a comprehensive analysis using DLBCL and other lymphoma cell lines, Chapuy and colleagues tested the efficacy of four BET inhibitors (JQ1, OTX015, I-BET151 and I-BET7762) in lymphoma cell lines in vitro and in xenograft models. Twenty DLBCL cell lines were tested, with molecular profiles that correspond to the genetic heterogeneity of the human disease. The four BET protein inhibitors performed with comparable potency and induced G1 cell cycle arrest in all B-cell lymphoma lines tested including Burkitt lymphoma (BL) cell lines. BL is an aggressive lymphoma characterized by t(8;14) resulting in MYC overexpression and MYC-dependent lymphomagenesis. JQ1 treatment of NSG mice transplanted with DLBCL cells resulted in lymphoma mass reduction and prolonged survival. The authors identified two groups of genes targeted by BET protein inhibitors in DLBCL cell lines. The first group appears common across DLBCL histological and molecular subtypes. It includes MYC and E2F transcriptional targets, but also members of the B-cell receptor (BCR) signalling and the MYD88/Toll ligand receptor (TLR) pathways. ChIP-seq and colocalization analysis identified super-enhancers which are predicted to control expression of transcription factors such as BCL6, PAX5 and IRF8, critical for the germinal centre (GC) programme and lymphomagenesis in DLBCL. A second group includes genes specific to histological subtypes, for instance the IRF4 gene in activated (ABC) but not in the GC B cell (GCB) subtype [Chapuy et al. 2013]. Specific to ABC DLBCL, another group identified NF-κB gene signatures in the targets of JQ1 and CPI203 inhibitors [Ceribelli et al. 2014].
A few studies have tested the potential synergy of BET inhibitors with other drugs, interestingly many of them in lymphomas. In a high-throughput drug screening JQ1 or CPI203 were tested in two-drug combinations with 466 drugs, which are either approved cancer therapies or in early phases of clinical development. A significant synergy of BET protein inhibitors with ibrutinib, a Bruton kinase inhibitor and with PI3 kinase and mTOR pathway inhibitors was documented, both in vitro and in vivo [Ceribelli et al. 2014].
CPI203 was shown to have a synergistic effect with lenalidomide in bortezomib-resistant mantle cell lymphoma (MCL), both in vitro in cell lines and in a xenograft model [Moros et al. 2014]. Plasmacytic differentiation, driven by IRF4 overexpression, has been linked to bortezomib resistance in MCL [Perez-Galan et al. 2011], while MYC is a direct IRF4 target in activated B cells and myeloma [Shaffer et al. 2008]. Combining CPI203 with lenalidomide inhibited transcriptional overexpression of IRF4 and MYC and overcame bortezomib resistance in vivo [Moros et al. 2014].
The most intriguing combination of BET inhibitors is probably with inhibitors of the enhancer of the zester homologue 2 (EZH2), one of the components of the polycomb-repressive complex 2 (PRC-2). EZH2 is a histone methyltransferase, responsible for the deposition of the repressive H3K27me3 epigenetic mark. In the developing GC, EZH2 generates repressive, bivalent chromatin domains at genes responsible for B-cell exit from the GC and mice with conditional deletion of EZH2 fail to develop GC. Somatic activating mutations or overexpression of EZH2 generates a differentiation block and together with BCL2 may drive lymphomagenesis [Beguelin et al. 2013; Heyn and Esteller, 2013]. EZH2 somatic mutations have been described in follicular lymphoma (FL) and DLBCL [Yap et al. 2011] and EZH2 inhibitors are currently in clinical development [Mccabe et al. 2012; Keilhack, 2013]. MYC represses the EZH2-targeting miR-29 and supports EZH2 overexpression, while EZH2 supresses the MYC-targeting miR-494, in a positive-feedback loop [Zhang et al. 2012b]. JQ1 disrupts the MYC-miRNA-EZH2 circuit and synergizes with the EZH2 inhibitor DZNep, to further inhibit MYC, restore suppressive miRNA function and reduce the clonogenic capacity of aggressive MCL and Burkitt-like cell lines in vitro [Zhao et al. 2013a].
In mature T-cell lymphomas, the preclinical evidence for BET inhibitors activity is limited, although this diseases group could be a favourable target, because of the documented therapeutic efficacy of the HDAC inhibitors vorinostat and panobinostat in some T-cell lymphomas. OTX015 effectively induces cell cycle arrest, but not apoptosis in five out of seven anaplastic large-cell T-cell lymphoma cell lines tested. MYC mRNA level is reduced in the sensitive cell lines, but further research is needed to define the role of BET inhibitors here [Boi et al. 2012].
Evidence of clinical efficacy was reported in the OTX015 dose-escalation phase I trial interim analysis, which included 11 lymphoma patients. Partial response as assessed by imaging was reported in 1 DLBCL and 1 lymphoplasmacytic lymphoma patient, after just two courses [Herait et al. 2013]. This early report supports further the clinical development of BET protein inhibitors in lymphoma.
Myeloproliferative neoplasms
The therapeutic potential of I-BET151 in JAK2V617F-positive myeloproliferative neoplasms (MPN) has been investigated in the erythroblastic cell line HEL and primary erythroid progenitor cells from patients with polycythaemia vera. I-BET151 inhibits expression of the LMO2 oncogene, a downstream target of JAK2 and important survival factor in MPN, leading to proliferation arrest, apoptosis and reduced clonogenic capacity in vitro. JAK2 inhibitors have an additive effect to I-BET151. Of note, I-BET151 had no adverse impact on normal erythroid progenitors [Wyspianska et al. 2014].
Conclusions
The BET protein inhibitors are the first successful example of pharmaceutical inhibition of epigenetic readers, a large and diverse group of epigenetic regulators. Their development has been facilitated by modern molecular technologies and advances in structural biology and medicinal chemistry and offers for the first time the opportunity to target cancer drivers, addictions and dependencies such a MYC, which for decades were thought to be ‘un-druggable’ (Figure 1).
Most of the BET protein inhibitors are structurally based on the familiar benzodiazepine template and have inherited their favourable pharmacological properties. There is compelling preclinical evidence of antitumour efficacy in refractory haematological malignancies particularly in acute leukaemia, myeloma and some lymphomas at drug levels that are achievable in vivo, with sufficient data to suggest acceptable off-target effects. Although little has been divulged so far from the ongoing early phase clinical trials, the results are encouraging and progress to phase II trials is anticipated. Unlike previous epigenetic therapies, pre-clinical research has already produced a vast amount of in-depth mechanistic knowledge, which can be harnessed in optimising their clinical application. Future research could include combinations with existing therapies and the development of biomarkers.
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.